
Abstract
In order to facilitate Health Canada’s study on background radiation levels in country foods, an in-house radio-analytical method has been developed for determination of polonium-210 (210Po) in fish samples. The method was validated by measurement of 210Po in a certified reference material. It was also evaluated by comparing 210Po concentrations in a number of fish samples by another method. The in-house method offers faster sample dissolution using an automated digestion system compared to currently used wet-ashing on a hot plate. It also utilizes pre-packed Sr-resin® cartridges for rapid and reproducible separation of 210Po versus time-consuming manually packed Sr-resin® columns.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A large fraction of the natural background radiation experienced by individuals through ingestion of food and water is from the radionuclide polonium-210 (210Po). 210Po delivers about 60 % of the annual effective dose received through the ingestion pathway from all naturally occuring radionuclides expect potassium-40 [1]. Fish products are the major contributor for dietary intake of 210Po when compared to different dietary components and their average annual consumption rates. Based on the reference concentration values of the radionuclides in fish products from the UNSCEAR 2000 report, 210Po constitutes more than 80 % of all long-lived radionuclides in the uranium and thorium series combined [1]. The affinity of 210Po for protein enables it to pass through the food chain, and increased body burdens of 210Po have been found where diets include protein-rich meat and seafood [2]. The radiation protection bureau (RPB) of Health Canada has undertaken a study for the measurement of 210Po in fish samples collected from Canadian lakes, rivers and oceans in order to determine the background concentration for this radionuclide. An accurate knowledge of the background concentration of 210Po in fish is important in order to enrich the existing data gaps to better reflect region-specific dietary intake information. For example, the current UNSCEAR report on the exposure from natural radiation sources contains the measurement data from only the United States for the concentration of 210Po in fish products in North America [1]. Background concentration data is also helpful in establishing guidelines for the threshold level of intake for food safety. Moreover, measurement of 210Po in fish and the associated radiological dose to human provides a perspective to compare and communicate radiological risk from consumption of fish following an accidental release of artificial radionuclides in the aquatic biota.
The most common features of the analytical methods developed and applied for determination of 210Po in environmental and biological samples are (a) dissolution of a representative sub-sample, (b) extraction and separation of 210Po, (c) source preparation and (d) quantitative measurement of 210Po using alpha spectrometry [3]. The purpose of sample dissolution is to mineralise the target radionuclides and render them in a chemical form suitable for subsequent use. For sample dissolution, wet-ashing has been shown to be the preferred method of choice over dry-ashing due to substantial loss of 210Po through volatilisation [3–7]. For alpha spectrometic measurement, a source preparation step is necessary in order to deposit polonium in a thin layer on a disk or a filter. Although source preparation can be conveniently carried out directly on a dissolved sample by spontaneous deposition of polonium on a silver disk without any extraction and separation, the presence of interfering ions can make the procedure less reliable both by reducing the deposition yield of polonium and increasing the thickness of the deposited layer. Thus the alpha spectra can be degraded, in extreme cases, even preventing the alpha spectrometric determination of the polonium [3, 8]. Extraction and separation of 210Po has been carried out by ion-exchange chromatography [9], solvent extraction [10–13], and extraction chromatography [8, 14–16]. Optimisation of the source preparation for alpha spectrometric determination of 210Po has been evaluated in a number of studies [17–21].
The RPB did not have a validated method for determination of 210Po in fish samples and contracted out the radiochemical analysis to commercial analytical service laboratories. An in-house method development was thus deemed to be important in terms of long term cost saving as well as having a better control over the quality assurance for the overall study. A review of the existing analytical methods suggested two possible areas for further improvisation, namely faster wet ashing and extraction chromatographic separation that will lead to an improved efficiency for determination of 210Po in fish samples. The details of the method development and validation are presented here.
Experimental
Sample digestion
In order to address the radiological concern from the dietary intake, the edible portion of the fish sample (flesh) was used for the measurement of 210Po. After separating from the skin and bone, the flesh was homogenised in a food processing blender and was kept in a freezer (−20 °C) until further analysis. For sample dissolution, a DigiPREP HT 250-10 digestion system (SCP Science, Baie D’Urfe, QC, Canada) has been used in this work. It consists of a graphite heating block that can accommodate ten tall cylindrical digestion tubes (300 mL, glass). The heating block contains ten built-in cylindrical cavities (5 cm depth) to house the bottom of the digestion tubes to provide a more uniform heating similar to a heating mantle. The digestion system was placed inside a chemical fume hood.
A 20 g aliquot of homogenised fish flesh was placed inside a pre-cleaned digestion tube with a few pre-cleaned boiling chips. A known quantity (50 mBq) of 209Po standard solution (Reference No. 1725-48, Eckert & Ziegler Isotope Products, Valencia, California, USA) was added as a tracer for chemical recovery. A 60 mL aliquot of concentrated nitric (70 %) acid was added to each digestion tube. The sample digestion was carried out in four pre-programmed steps of time-to-temperature and time-at-temperature conditions. In the first step, the temperature of the heating block was raised from ambient to 180 °C (in 30 min) and was held at 180 °C for 1 min. During this heating step, if excessive foaming was observed, the digestion tube was removed from the heating block, gently swirled to subside the foaming and placed back in the heating block. When the frothing subsided, a glass ball condenser (SCP Science, Baie D’Urfe, QC, Canada) was placed on top of the digestion tube to efficiently cool the vapour rising into the glass finger tips of the condenser during the digestion and to allow the condensed acid to flow back into the digestion tube for efficient refluxing. In the second step the temperature was lowered to 140 °C (in 10 min) and was held for 2 h. In the third step, the temperature was raised to 240 °C (in 15 min) and was held for 2 h. In this step, 10 min after the temperature was raised to 240 °C, the scaffold (a Teflon coated aluminum rack that holds the digestion tubes) was raised from the heating block and 12.5 mL of hydrogen peroxide was added (drop wise) to the digestion tube. Once the fizzing subsided, the scaffold was lowered to replace the digestion tube back in the heating block. Addition of hydrogen peroxide was repeated three more times in 20 min time intervals. In the fourth step the temperature was lowered to 140 °C (in 15 min) and was held for 1 h. When the temperature reached 140 °C, the glass ball condenser was removed from the digestion tube, the solution was allowed to evaporate to 5 mL, and the digestion tube was removed from the heating block. The solution of the digested fish sample was transferred to a 50 mL Teflon beaker. The digestion tube was rinsed three times with 10 mL of 2 M hydrochloric acid to quantitatively transfer the content to the Teflon beaker. To carry out the extraction chromatographic separation of 210Po, the digested sample solution was converted from the nitric acid to hydrochloric acid (2 M) medium as described below.
The beaker was placed on a hot plate to evaporate the solution at 90 °C. When the volume of the solution was reduced to about 5 mL, a 5 mL aliquot of concentrated hydrochloric acid was added to the beaker and the heating was continued until approximately 1 mL of the solution was left. This was repeated three times, each time, by addition of a 5 mL aliquot of 2 M hydrochloric acid and evaporation to approximately 1 mL. This solution was transferred to a 50 mL polypropylene tube and the beaker was rinsed thoroughly with 2 M hydrochloric acid; the final volume in the tube was adjusted to 30 mL.
Extraction chromatographic separation
Separation of 210Po from the digested sample matrix containing 210Pb and 210Bi was carried out by extraction chromatography using a Sr-resin® cartridge as described in the Eichrom method for 210Pb and 210Po in water [22] except for stripping of 210Pb. 210Pb was stripped with 20 mL of 0.05 M ammonium citrate.
Measurement of 210Pb-210Bi-210Po by liquid scintillation counting
For the initial chromatographic separation, a liquid scintilltion counter (Tri-Carb 3180TR/SL, PerkinElmer Inc., Woodbridge, ON, Canada) was used for the measurement of 210Pb-210Bi-210Po in the load, rinse and the strip fractions. In order to determine the appropriate pulse decay discriminator (PDD) settings for alpha–beta separation in various load, rinse and strip fractions, a PDD settings optimisation was carried out using a alpha emitting radionuclide standard (210Po, 1000 Bq) and a beta emitting radionuclide standard (90Sr/90Y, 1000 Bq) prepared in 2 M HCl, 1 M HNO3, 0.1 M HNO3, and 0.05 M ammonium citrate. A 1 mL aliquot of each of the load, rinse, and strip fractions was mixed with 19 mL of liquid scintillation cocktail (OptiPhase HiSafe 3, PerkinElmer Inc., Woodbridge, ON, Canada) and counted for 30 min on the liquid scintillation counter (LSC) with the optimised PDD setting for alpha–beta discrimination.
Deposition of 210Po on silver disk and alpha spectrometric measurement
The stripped solution containing 210Po was transferred to a 50 mL Teflon beaker. The tube was rinsed three times with 5 mL of 2 M HCl to quantitatively transfer the content to the Teflon beaker. The conversion procedure from HNO3 to HCl medium was repeated as described in the sample digestion section (above) except in the final step a 30 mL aliquot of deionised water was used instead of 2 M HCl and the solution was transferred to a pre-cleaned 50 mL glass beaker containing a magnetic stir bar (Teflon coated). The beaker was placed on a hot plate with stirring (magnetic) capability. 0.33 g of ascorbic acid was added to the beaker and dissolved by magnetic stirring. The pH of the solution was adjusted to 1 using 2 M HCl. For auto deposition of 210Po, preparation of a silver disk and its mounting inside the solution in the beaker was carried out as described in the Eichrom method [22]. The deposition of 210Po on the silver disk was carried out for 3 h at 90 ± 5 °C with moderate stirring. After deposition the silver disk was cleaned with deionised water, rinsed with ethanol (non-painted side) and was dried under an infra-red heat lamp.
The deposited 210Po on the silver disk was measured using a bench top Alpha Analyst system (Canberra Industries Inc., Meriden, CT, USA), where the alpha particles were detected using 25 mm silicon PIPS detectors. The samples were placed on the second shelf, about 2 cm away from the detector, and counted for 24 h using the recoil suppression mode. 210Po activity was then calculated using the APEX-Alpha software using 209Po standard as a yield tracer.
Results and discussion
Sample dissolution by wet ashing
Wet ashing is typically carried out by repetitive digestion on a hot plate in the presence of concentrated acids and hydrogen peroxide in an open beaker (often with a lid to prevent fast evaporation of acids) [3]. Conventional digestion on a hot plate is slow and requires close attention to avoid the risk of cross contamination, formation of insoluble salts, exposure to acid fumes and corrosion and charring during evaporation [23]. Wet ashing carried out on a hot plate can be quite time consuming, taking up to several days to completely dissolve a biological sample such as meat or fish especially if it contains fat [24–26]. Although microwave digestion can be an attractive alternative to open beaker digestion, it can only handle a very small amount of organic sample (typically, 0.5 g dry and 2–3 g for wet sample) per digestion vessel and thus a larger sample (20–25 g, wet weight) may need to be split into a number of digestion vessels to achieve the required sensitivity for the measurement of low levels of 210Po in fish samples [23, 27]. This also leads to an increase in digestion time that will lower the sample throughput capacity. The digestion system (DigiPREP HT 250-10) used in this work was found to efficiently dissolve a fish flesh sample (20 g, wet weight) into a clear liquid solution in less than 7 h without any visible fat residue (up cooling) at the end of the digestion. Tall cylindrical sample digestion tubes, uniform heating inside the built-in ceramic cavities, programmable multistage temperature control, and glass ball condensers to cool and condense the acid vapour to flow back in the digestion tubes for efficient refluxing are some of the features of the automated digestion system that enabled a more time efficient dissolution compared to time consuming conventional open beaker digestion for a similar size (20 g, wet weight) sample. Simultaneous digestion of up to 10 samples within a compact system inside the fume hood is also another attractive feature to provide improved sample throughput.
Optimisation of PDD setting for alpha/beta discrimination
For initial optimisation of the extraction chromatographic separation, liquid scintillation counting was used for the measurement of 210Pb, 210Bi, and 210Po in the load, rinse and strip fractions. In liquid scintillation counting, the pulse height spectrum of an alpha particle often overlaps with that of a beta particle. However, the decay time of light produced by an alpha particle in a suitable liquid scintillator is longer than that of the light produced by a beta particle. Discrimination of liquid scintillation signals resulting from alpha and beta emitting radionuclides can be carried out by the difference in the lengths of the pulse decay events [28]. Table 1 shows the data for discrimination of alpha and beta pulses (expressed as % spillover) at various PDD settings.
The 210Po and 90Sr/90Y standards were prepared in 2 M HCl, 1 M HNO3, 0.1 M HNO3, and 0.05 M ammonium citrate in order to match the chemical composition of the load, rinse and the strip fractions to account for the quenching effects. A curve of the % spillover versus the PDD settings was constructed for both the 210Po and 90Sr/90Y standards. The optimum PDD setting for α/β discrimination is where the two spillover curves intersect. The optimum PDD settings for α/β discrimination in 2 M HCl, 1 M HNO3, 0.1 M HNO3, and 0.05 M ammonium citrate were 151, 148, 157, and 158, respectively. Since the PDD setting for α/β discrimination in 0.1 M NHO3 and 0.05 M ammonium citrate were close to each other, a PDD setting of 157 was used for the measurement in both conditions.
Extraction chromatographic separation of 210Pb-210Bi-210Po
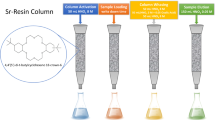
Sr-Resin® has been widely used for extraction chromatographic separation of 210Po from 210Pb, 210Bi and other interfering radionuclides if present in a sample [8, 29–32]. Typically, a digested sample solution is converted to 2 M HCl before loading on the Sr-Resin®. 210Pb and 210Po is retained on the column while 210Bi and other interfering radionuclides are eluted during the loading (2 M HCl) and rinse (using 2 M HCl) steps [8, 31]. 210Po and 210Pb are stripped from the Sr-Resin® using 6 M HNO3 and 6 M HCl, respectively. As compared to more concentrated stripping reagents (6 M HMO3 and 6 M HCl), milder reagents have been previously used for stripping 210Po (1 M HNO3 and 0.1 M HNO3) and 210Pb (0.05 M ammonium citrate or water) for determination of these radionuclides in water samples [22, 33]. Since the milder reagents would be less corrosive and would also lead to cost saving, 1 M HNO3, 0.1 M HNO3, and 0.05 M ammonium citrate were investigated in this work as stripping reagents for the separation of 210Po and 210Pb from digested fish samples. Figure 1a shows the separation of a standard mixture of 210Pb-210Bi-210Po (4 Bq; radionuclides are in secular equilibrium) prepared in deionised water in the presence of 2 M HCl.

Extraction chromatographic separation of 210Pb-210Bi-210Po using Sr-Resin® cartridge, a deionised water spiked with 210Pb-210Bi-210Po (4 Bq), b fish sample spiked with 210Pb-210Bi-210Po (4 Bq); LD load (digested sample after chloride conversion, in 2 M HCl), RF1 and RF2 rinse fractions (5 mL of 2 M HCl); Po1 polonium strip solution 1 (5 mL of 1 M HNO3); Po2, Po3, and Po4 polonium strip solutions (5 mL of 0.1 M HNO3), Pb1, Pb2, Pb3, and Pb4 lead strip solutions (5 mL of 0.05 M ammonium citrate)
As anticipated, 210Bi was not retained on Sr-Resin® and was eluted during the loading and rinse steps. According to the separation conditions as described in the Eichrom analytical procedure [22], recovery of 210Po (in 5 mL of 1 M HNO3 and 10 mL of 0.1 M HNO3) was about 90 % and recovery of 210Pb (in 10 mL of 0.05 M ammonium citrate) was quantitative (~100 %). Also, approximately 10 % of the spiked 210Po was observed in the first 5 mL fraction of 0.05 M ammonium citrate (stripping reagent for 210Pb). To increase the 210Po recovery, the stripping volume of 0.1 M HNO3 was increased from 10 mL (as described in the Eichrom analytical procedure) to 15 mL. As shown in Fig. 1a, this increased the 210Po recovery (in 5 mL of 1 M HNO3 and 15 mL of 0.1 M HNO3) to about 95 % while reducing the concentration of 210Po in 0.05 M ammonium citrate to approximately 5 %. A further increase in volume of 0.1 M HNO3 did not improve the recovery of 210Po.
In order to assess the application of the extraction chromatographic separation in the presence of a fish matrix, homogenized flesh samples from three fish (20 g, wet weight each) were spiked with a standard mixture of 210Pb-210Bi-210Po (4 Bq per sample) and digested according to the procedure described above. The elution profile for the extraction chromatographic separation of 210Pb, 210Bi, and 210Po in a digested fish matrix is shown in Fig. 1b. The elution profile of 210Po was similar to that observed in deionised water. The spike recovery of 210Po (in 5 mL of 1 M HNO3 and 15 mL of 0.1 M HNO3) in the digested fish samples was found to be 85 ± 5 % (average ± 1-standard deviation, n = 3). Lower recovery of spiked 210Po in the digested fish samples (compared to the spiked standard in deionized water) was likely due to the evaporative loss during digestion at elevated temperature. The elution profile of 210Pb for the digested fish sample was significantly different. No 210Pb was observed in the first 5 mL fraction of 0.05 M ammonium citrate and <12 % of the spiked 210Pb was recovered in the second 5 mL fraction (of 0.05 M ammonium citrate). In order to achieve quantitative recovery of 210Pb, the strip volume of 0.05 M ammonium citrate needed to be increased from 10 mL (as described in the Eichrom procedure [22]) to 20 mL (Fig. 1b). After this modification, spike recovery of 210Pb increased to 92 ± 4 %. The change of elution profile for 210Pb is likely due to the matrix effect of the digested fish sample resulting from the residual organic and inorganic components present in the loading solution. Interestingly, in the digested fish samples, the spike recovery of 210Bi in the loading and strip fractions was 111 ± 6 %. The higher (>100 %) recovery could be due to the interference contribution of 40K (present in the fish sample) to the 210Bi liquid scintillation signal.
Determination of 210Po in fish samples: method validation
Although it was used as a convenient radionuclide detection system for the optimisation of extraction chromatographic separation, the liquid scintillation counting could not be used for the measurement of 210Po natively present in the fish samples due to lack of the required detection limit. Alpha spectrometry was used for the determination of 210Po in the fish samples. For the alpha spectrometric determination, the overall chemical recovery of polonium (as determined by the 209Po yield tracer) was 76 ± 3 %. It was lower than the recovery of 210Po (85 ± 5 %) obtained when a number of fish samples were spiked with known quantity (4 Bq) and were determined by liquid scintillation counting after wet digestion and extraction chromatographic separation. The decrease in chemical recovery for alpha spectrometric determination was likely due to the additional evaporative loss during the second chloride conversion step before spontaneous deposition of polonium on silver disk. Shorter auto deposition time (3 h) was also a contributing factor for lower chemical recovery. Lower chemical recovery of polonium (50–70 %) was also reported by Vajda et al. when sample dissolution was carried out by wet ashing through repetitive digestion on a hot plate in the presence of concentrated acids (HNO3 and HCl) and hydrogen peroxide [8].
A certified reference material (IAEA-414) purchase from the International Atomic Energy Agency (IAEA) for radionuclides in mixed fish from the Irish Sea and North Sea was used to validate the method for determination of 210Po. IAEA-414 provides an information value (2.1 Bq/kg) for 210Po (present in secular equilibrium with 210Pb) with a 95 % confidence interval (1.8–2.5 Bq/kg) for a reference date of January 1, 1997. 5 g (dry weight) of the standard reference material (IAEA-414) was analysed in triplicate. Since the average moisture content of the flesh portion for a fresh fish is about 75 % and the digestion procedure was optimised for a 20 g (wet weight) aliquot of a homogenized fish (flesh portion), 5 g of dry weight for IAEA-414 was a reasonable amount for the method validation. The measured value of 210Po in the IAEA-414 was found to be 1.9 ± 0.4 Bq/kg (average ± 2 standard deviation). The relative bias between the information value (valueIAEA) and the measured value (valueanalyst) for 210Po concentration was less than 10 %. For performance evaluation, the Z—score and the U—score values were calculated according to the IAEA’s HELCOM-MORS proficiency test [34]. The HELCOM-MORS proficiency test (PT) was organized by the IAEA’s Marine Environmental Laboratory to evaluate the analytical performance for radionuclides on a fish flesh sample (IAEA-414) for the participating laboratories. Normally, the reference value of an analyte in a certified reference material is used for the Z—score and U—test. However, since there is not any suitable reference material associated with fish matrix and the reference value of 210Po, Z—score and U—test were carried out using the information value of 210Po from IAEA-414 certificate. The Z—score was calculated according to Eq. 1 as shown below:
As outlined in the PT guidance, on the basis of the “fitness for purpose” principle, the target value of the standard deviation (σ) is: σ = 0.1 × valueIAEA.
A laboratory performance is evaluated as satisfactory if \( \left| {Z - {\text{score}}} \right| \le 2 \); questionable for \( 2 \le \left| {Z - {\text{score}}} \right| \le 3 \), and unsatisfactory for \( \left| {Z - {\text{score}}} \right| \ge 3 \).
The value of the U—score, expressed as, \( U_{\text{test}} \) was calculated according to (Eq. 2) as follows:
where \( {\text{Unc}}_{\text{IAEA}} \) and \( {\text{Unc}}_{\text{Analyst}} \) are expanded uncertainties (2σ), which cover 95 % confidence interval. The limiting value of the U—score for the PT was set to be 2.58, so that a laboratory passes the test if \( U_{\text{test}} < 2.58 \).
The values for the Z—score and U—score for the current method were 0.95 and 0.24, respectively. Thus, the method presented here, passes the performance criteria for both Z—score and U—score according to the IAEA’s HELCOM-MORS proficiency test.
Determination of 210Po in fish samples: method intercomparison
Prior to the development of this method, RPB used to contract out the measurement for 210Po to Saskatoon Research Council (SRC), a commercial analytical service laboratory. A method intercomparison was undertaken to further evaluate the method performance on wet fish flesh samples. A number of homogenized fish flesh samples were split into two aliquots. One set of these samples was analyzed by the SRC and the second set was analyzed by the method described in this paper. The results for the 210Po concentration in these fish samples analyzed by the two laboratories are shown in Table 2. For the measurements carried out in RPB, concentration of 210Po in a fish sample and the associated combined standard uncertainty were calculated according to the procedure described in the MARLAP manual [35].
Comparison of the two methods was carried out by means of Z—score and Chi squared (\( \chi^{2} ) \) test as described by Saidou et al. [36]. Since the concentration of 210Po in a fish sample is not known, Saido et al. [36] used following equation (Eq. 3) to calculate the Z—score value:
The Chi squared (\( \chi^{2} ) \) value was calculated using the equation (Eq. 4) as shown below:
The \( \left| {Z - {\text{score}}} \right| \) values for each pair of measurement by the two methods were <2. A \( \left| {Z - {\text{score}}} \right| \) value of <2 indicates that there is no significant difference between the two measured values of 210Po for a fish sample within 95 % confidence interval. The concordance of the two methods was evaluated by a \( \chi^{2} \) test for measurements of multiple samples [36]. For measurement of 12 fish samples by the two methods, the result of the \( \chi^{2} \) test on the Z—score values was 15.3. It is lower than the critical value (21.0) for 95 % confidence interval (α = 0.05) and 12 measurements (n = 12). This indicates that there is no significant difference between the two methods within a 95 % confidence interval.
Minimum detectable concentration and sample throughput
Minimum detectable concentration (MDC) was calculated according to the MARLAP recommendations [35] using the Poisson-Normal approximation with Type I (false positive) and Type II (false negative) error rates at 5 % (α = β = 0.05). The MDC for 210Po was 0.1 Bq/kg for the measurement of a 20 g aliquot of a fish flesh sample with a counting time of 24 h using an alpha spectrometer. The SRC reported a sample specific MDC that ranged from 0.2 to 0.3 Bq/kg. Data available from the UNSCEAR 2000 report for the measured concentration of 210Po in fish products from the United States range from 0.15 to 55 Bq/kg [1]. The MDC for the in-house method was, thus, considered to be adequate in terms of the required sensitivity for measurement of the background level of 210Po in Canadian fish samples. The current sample throughput of the in-house method is eight fish samples in a week (37.5 working hours) by one laboratory personnel using two chemical fume hoods and one alpha spectrometer with eight detector chambers.
Conclusions
An in-house radio-analytical method has been developed for the determination of 210Po in fish samples to facilitate Health Canada’s study on background radiation levels in country foods. Over the existing analytical methods for determination of 210Po in fish samples, this the new method offers improvisations in two different aspects: (1) faster sample dissolution (in <7 h) using an automated digestion system compared to wet ashing on a hot plate (in several days), and (2) rapid and reproducible extraction chromatographic separation of 210Po using a standardized (2 mL) pre-packed Sr-resin® cartridge over a manually packed Sr-resin® column that requires additional packing time. A certified reference material (IAEA-414) for radionuclides in a mixed fish sample was used to validate the method for determination of 210Po. The relative bias between the measured concentration and IAEA’s information value was less than 10 %. The method also passed the performance criteria for both Z—score and U—score for determination of 210Po in the certified reference material according to the IAEA’s HELCOM-MORS proficiency test. Further, an intercomparison study on 12 fish flesh samples between the in-house method and a method used by the SRC demonstrated no significant difference on the measurement results for 210Po concentration within the 95 % confidence interval on the basis of Z—score values and a Chi squared (\( \chi^{2} ) \) test. The MDC (0.1 Bq/kg) of the in-house method is adequate in terms of the required sensitivity for measurement of the background level of 210Po in Canadian fish samples. Since the completion of the method development, over 100 fish samples have been analysed for 210Po for the Canadian background radioactivity measurement study and will be presented in a separate publication.
References
UNSCEAR (2000) sources and effects of ionizing radiation. United Nations Scientific Committee on the Effects of Atomic Radiation UNSCEAR 2000, report to the general assembly with scientific annexe, United Nations, New York
Alam A, Mohamed CAR (2011) Int Food Res J 18:1–10
Matthews KM, Kim CK, Martin P (2007) Appl Radiat Isot 65:267–279
Seiner BN, Morley SM, Beacham TA, Hanley MM, Gregory S, Metz L (2014) J Radioanal Nucl Chem 302:673–678
Martin A, Blanchard RL (1969) Analyst 94:441–446
Cleary JJ, Hamilton EI (1968) Analyst 93:235–236
Mabuchi HJ (1963) Inorg Nucl Chem 25:657–660
Vajda N, LaRosa J, Zeisler R, Danesi P, Kis-Benedek GY (1997) J Environ Radioact 37:355–372
Pacer RA (1983) J Radioanal Chem 77:19–28
Uesugi M, Noguchi M, Yokoyama A, Nakanishi T (2010) J Radioanal Nucl Chem 283:577–584
Kim C-K, Lee MH, Martin P (2009) J Radioanal Nucl Chem 279:639–646
Chen Q, Hou X, Dahlgaard H, Nielsen SP, Aarkrog A (2001) J Radioanal Nucl Chem 249:587–593
Veronneau C, Aupiais J, Dacheux N (2000) Anal Chim Acta 415:229–238
Maxwell SL, Culligan BK, Hutchison JB, Utsey RC, McAlister DR (2013) J Radioanal Nucl Chem 298:1977–1989
Reischmann FJ, Trautmann N, Herrmann G (1984) Radiochim Acta 36:139–143
Ordonez-Regil E, Iturbe JL (1993) J Radioanal Nucl Chem 17:47–53
Lee HM, Hong GH, Baskaran M, Kim SH, Kim YI (2014) Appl Radiat Isot 90:170–176
Porntepkasemsan B, Srisuksawad K, O-manee A TIChE International Conference 2011, Hatyai, Songkhla Thailand. http://www.chem.eng.psu.ac.th/tiche2011/TCHE/data/paper/international/ms/poster/ms006.pdf. Accessed 07 May 2015
Lee MH, Lee CH, Song K, Kim CK, Martin P (2010) Bull Korean Chem Soc 31:2488–2492
Henricsson F, Ranebo Y, Holm E, Roos P (2011) J Environ Radioact 102:415–419
Vesterbacka P, Ikaheimonen TK (2005) Anal Chim Acta 545:252–261
Lead-210 and polonium-210 in water. Analytical procedures. Eichrom Technologies LLC: Rev. 2.0. (OTW01). http://www.eichrom.com/docs/methods/pdf/otw01-20_pb-po-water.pdf. Accessed 15 April 2015
Louw I, Faanhot A, Kotze D (2009) Radioprotection 44:89–95
Clayton RF, Bradley EJ (1995) Sci Total Environ 173(174):23–28
Ham GJ, Ewers LW, Clayton RF (1997) J Radioanal Nucl Chem 226:61–65
Samad OE, Baydoun R, Jeaid HE (2010) Leban Sci J 11:39–45
Gjelsvik R, Brown J, Holm E, Roos P, Saxen R, Outola I (2012) Results from the NKS project GAPRAD (Filling knowledge gaps in radiation protection methodologies for non-human biota), Sralevern Rapport 2012:3. Østerås: Norwegian Radiation Protection Authority. http://www.nrpa.no/dav/a7950bbba1.pdf. Accessed 07 May 2015
L’Annunziata MF, Kessler MJ (2003) In: L’Annunziata MF (ed) Handbook of radioactivity analysis, 2nd edn. Academic Press, California
Miura T, Hayano K, Nakayama K (1999) Anal Sci 15:23–28
Biggin CD, Cook GT, MacKenzie AB, Pates JM (2002) Anal Chem 74:671–677
Vrecek P, Benedik L, Pihlar B (2004) Appl Radiat Isot 60:717–723
Lin Z, Wu Z (2009) Appl Radiat Isot 67:907–912
Johansson LY (2008) Determination of Pb-210 and Po-210 in aqueous environmental samples. Ph. D. Thesis. http://pbadupws.nrc.gov/docs/ML1105/ML110560301.pdf. Accessed 15 April 2015
HELCOM-MORS Proficiency Test, Determination of Radionuclides in Fish Flesh Sample, IAEA Analytical Quality in Nuclear Application No. IAEA/AQ/13, International Atomic Energy, Vienna, 2010
Multi-Agency Radiological Laboratory Analytical Protocols (MARLAP) Manual, NUREG-1576, EPA 402-B-04-001C, NTIS PB2004-105421, July, 2004
Saidou, Bochud F, Laedermann J-P, Njock MGK, Froidevaux P (2008) Appl Radiat Isot 66:215–222
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sadi, B.B., Chen, J., Kochermin, V. et al. A faster sample preparation method for determination of polonium-210 in fish. J Radioanal Nucl Chem 308, 843–850 (2016). https://doi.org/10.1007/s10967-015-4575-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-015-4575-6