
Abstract
The primary reaction products and reaction mechanism of uranium with oxygen were discussed from MP2 method with the relativistic core potential of SDD basis set for U and 6-311+G* for O. The molecular geometries, electronic structure and energies of uranium oxides were obtained. The inspection on the three-dimensional potential energy surfaces of the U–O2 interaction suggested that the abstraction and insertion mechanism were responsible for the studied reactions. The abstraction reaction channel resulting in the formation of UO and O is favored because the energy barrier is remarkably smaller than the one of the insertion channel resulting in the linear OUO product directly.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As is known, there is a wide application perspective of actinide compounds although actinide chemistry poses a formidable challenge for chemists [1]. Uranium is the heaviest naturally occurring actinide element and largely known due to its use as a nuclear reactor fuel [2]. The reactivity of the uranium compounds is strongly influenced by the involvement of 5f electrons in chemical bonding. They are not easy to handle in the laboratory, so many electronic and geometric structures and related properties from experimental methods are limited [3–5]. On the other hand, they play important roles in advanced nuclear fuel cycles. Sparked by the above issues, a large number of theoretical calculations have been reported on uranium compounds [6–8]. In a few studies, basic bulk properties of actinide nitrides with emphasis on elastic and magnetic properties were considered. Some efforts have also been made to elucidate the bulk and surface properties of uranium. We recently reported theoretical investigations of interatomic potential energy surface (PES) of diatomic uranium [9].
Comprehensive experimental reviews are available on studying the uranium oxides at different circumstances [10, 11]. A variety of surface analysis techniques were utilized to study initial oxidation of uranium by water vapor including early stages of oxidation at the room temperature and the low temperature regimes. Considerable experimental efforts have also been devoted to studying the oxidation kinetic property of uranium metal [12, 13]. The results show that oxygen molecule on the clean uranium dissociated rapidly, reacted to form oxide with exterior uranium atom. The activation energy of oxidation is obtained to be about 46.0 kJ mol−1. Thermodynamic assessment of the uranium–oxygen system and the determination of the composition range of uranium oxides have been determined using thermogravimetric, X-ray diffraction and electrochemical techniques. The experimental work on the oxidation reaction and corrosion resistance of uranium has been recently reported [14, 15].
In contrast to the multiple experimental surveys on the uranium–oxygen reaction system, relatively few quantum chemical computations on the reaction have been reported so far. For convenience, the ab initio method is a very powerful theoretical technique for computing the molecular structure and some properties [16]. Uranium occupies a central position in the early actinide series, with only three 5f electrons hybridizing with the 6d and 7s electrons showing remarkable relativistic effective. Stuttgart/Dresden effective core potential (SDD) can successfully compensate the relativistic effect [17]. In this work, the MP2 method with the SDD basis set was employed to study the reaction of uranium with oxygen. The molecular geometries, the electronic structure and energies of the primary reaction products were obtained. The three-dimensional PESs of the reactions were studied to explore the reaction mechanism of uranium with O2. The present results elucidated the mechanism of the title reaction and further experimental investigation of the reaction.
Computational details
Geometries optimization of some uranium oxides were conducted without any constraint. Each optimized structure was confirmed by the frequency calculation to be the real minimum without any imaginary vibration frequency. The geometrical parameters, the frontier orbital energy (E HOMO, E LUMO), energy gap (ΔE = E LUMO − E HOMO), total energy (E 0), vibration frequencies (ν) and and IR intensities (I) were obtained.
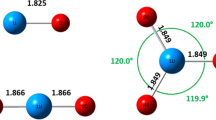
To explore the reaction mechanism of the U–O2 reaction system, some three-dimensional PESs based on the single point energy calculations are also constructed. In the case of three atoms system, it is well-known that the coordinate number of potential energy profile is (3 + 1) [18]. In the four-dimensional reference frame, if one of the four references such as angle is fixed, a three-dimensional PES will be obtained. Also, a three-dimensional PES with a fixed reaction angle can render a clear picture of diverse reaction modes. As can be seen from Fig. 1, α represents the angle of R UO and R OO, and β represents the angle of R UM and R MO, which the point M is the median of O–O length.

The coordinate of U–O system (M is the median of O–O length)
A non-uniform direct product grid in the internal coordinates was selected for the calculation of the PES. The PES for the reaction pathway was calculated by varying the bond length R UO from 0.17 to 0.42 nm, the bond length R OO from 0.11 to 0.36 nm, which gives a total of about 5,100 single points.
All the calculations were implemented at second-order perturbation theory (MP2) level with the relativistic effective core potential (ECP) of basis sets (SDD) for U and the 6-311+G* basis set for O. Computations were carried out with the Gaussian 03 program package [19].
Results and discussion
Geometrical and electric structure
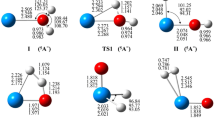
As we know, earlier experimental studies only reported the structure parameters of solid uranium oxide crystal. There is a lack of experimental data on the geometrical structure of gaseous oxides. Some data from theoretical literatures [20, 21] were employed to corroborate our present findings. With regard to the reaction of the uranium with oxygen, it is apparent that the primary reaction products are UO or UO2. The optimized geometrical parameters are shown in Table 1. On the basis of data, it can be concluded that the UO2 molecules including two conformers OUO and UOO. Both of them are linear molecules. The optimized U–O bond lengths for UO, OUO and UOO are 0.1812, 0.1806 and 0.1855 nm, respectively, which are in acceptable agreement with data from literatures [20, 21].
Meanwhile, the electronic characteristics of UO, OUO and UOO were summarized in Table 2. Judging by the frontier orbital energy gap (ΔE = E LUMO − E HOMO), the relative stability of an individual molecule in the gas phase can be determined. The larger energy gap (ΔE), which is resulted from higher energy of LUMO and lower energy of HOMO, indicates that neither losing nor capturing electron would happen on the title compound easily, so it is more stable. It is apparently that the OUO is more stable than UOO because the calculated ΔE of OUO (0.2715 a.u.) is larger than that of UOO (0.2404 a.u.). Same conclusion can be drawn from a quantitative viewpoint of total energy (E 0), which was also given in Table 2. Our MP2 calculations predicting vibration frequencies (ν) are rather consistent with the former theoretical calculations [20].
Potential energy surface
The uranium–oxygen system is one of the most complex metal oxide systems due to the high reactivity of uranium with oxygen. The imaginable products are UO or UO2. The diverse reaction manners of uranium with oxygen molecule can be simulated by the three-dimensional PES when the angle α is initialized at a fixed value of 180, 135, 90, and 45°, respectively. In Fig. 1a, it can be seen that as α is set to be 45°, the OUO molecule may immediately come into being via the U atom inserting into the O–O bond directly. The reaction manner can be precisely described when the angle β was set to be 90° in Fig. 1b.
Figure 2 displayed the PES for reaction of uranium with oxygen at α = 180°. As expected, a minimum pathway appeared in the smooth shape of the PES. At the initial step along the minimum pathway, the U–O distance reduced from 0.42 to 0.185 nm to form the intermediate UOO. In the meantime, the reaction potential energy decreased. Because the potential energy is 126.944 kJ mol−1 below the reactants, this step proceeds easily. The predicted bond distances of 0.185 nm (U–O) and 0.150 nm (O–O) of the intermediate UOO were in good agreement with the values of 0.1855 and 0.1550 nm in Table 1. In the next step, the O atom gradually removed because of space effect coming from U atom. The small energy barrier of about 42.917 kJ mol−1 makes the removed step energetically possible. Based on the above analysis, it can be assumed that a uranium atom abstracted an oxygen atom from an oxygen molecule to form UO. Thus, a direct abstraction mechanism for this reaction manner would be proposed.

The potential energy surface for the reaction of U with O2 at α = 180°
The three-dimensional PES of uranium–oxygen system at β = 90° was shown in Fig. 3 to illustrate the reaction mechanism of producing OUO directly from U + O2 reaction. In the minimum reaction pathway, when uranium atom moves forward to the median of O–O bond, the potential energy of reaction system also decreases. Consequently, the potential minimum located at R UO = 0.280 nm and R OO = 0.140 nm is visible, which is similar to the phenomenon in Fig. 2. In this stage, the O–O bond length gradually increases from 0.11 to 0.14 nm, revealing that the O–O bond is activated. In the next step, the O–O bond continuously increases to 0.36 nm till the distance between uranium and median of O–O bond length falls into zero. In such case, the OUO is formed and the proposed mechanism for reaction U + O2 → OUO is an insertion mechanism.

The potential energy surface for the reaction of U with O2 at β = 90°
To make more accurate comparison between the abstraction mechanism and the insertion mechanism, the potential energy diagrams along the minimum pathway for uranium interacting with oxygen molecule in different directions were plotted in Fig. 4. Because the three-dimensional PESs of α = 135° and α = 90° are extremely close to the one of α = 180°, we only displayed their potential energy diagrams in Fig. 4. Also the abstraction mechanism was applicable in the two reaction modes.

The potential energy diagram for the reaction of U with O2
In the abstraction reaction process, there is a potential energy barrier with respect to the minimal potential energy when a uranium atom binds to an oxygen molecule. As can be seen in Fig. 4, the small potential energy barrier corresponds to the least energy requiring for the cleavage of O–O bond in UOO, which makes the O–O bond fission very easy.
Upon examining the potential energy diagram at β = 90°, there is a minimal and maximal potential energy throughout the insertion reaction pathway. The high energy barrier of about 107.340 kJ mol−1 resulting from energy difference between the minimal and maximal potential energy represents activation barrier of the formation OUO insertion product.
According to the potential energy diagrams and reaction energy barrier for the U + O2 reaction, it is apparent that the abstraction channel is energetically favorable since the energy barrier is significantly lower than that of insertion channel. Our theoretical findings are in good consistent with previous experimental investigations [6, 7] that O2 rapidly dissociated and reacted with uranium atom when O2 molecules were absorbed in the surface of uranium metal.
Conclusion
MP2 calculations in combination with ECP of SDD and 6-311+G* basis sets were applied to study the primary oxidation process of uranium surface. The geometrical parameters, frontier orbital energy gap (ΔE = E LUMO − E HOMO), and the vibrational frequencies together with IR intensities of some uranium oxides were also investigated at the same theoretical level. The three-dimensional PESs of about 5,100 single point energy for uranium–oxygen reaction system are presented. Based on analyzing the characteristics of different minimum reaction pathways, the abstraction and insertion mechanism are proposed. The abstraction mechanism shows that the reaction is most likely to proceed through the following steps: U + O2 → UOO → UO + O, which is found to be energy barrier of 42.917 kJ mol−1. While the insertion mechanism indicates that the U immediately inserts into an O–O bond resulting in a linear OUO insertion product with an energy barrier of 107.340 kJ mol−1. As a consequence, the abstraction reaction channel is energetically predominant over the insertion reaction channel. The present work made a contribution to explain the experimental observations because the abstraction reaction was in agreement with the experimental findings.
References
Morss LR, Edelstein NM, Fuger J (2006) The Chemistry of the actinide and transactinide elements. Springer, New York
Dholabhai PP, Ray AK (2007) J Alloy Compd 444–445:356–362
Amayri S, Reich T, Arnold T (2005) J Solid State Chem 178:567–577
Fujimori S, Terai K, Takeda Y (2006) Phys B Condens Matter 378:995–996
Soto-Guerrero J, Gajdošová D, Havel J (2001) J Radioanal Nucl Chem 249:139–143
Roose BO, Gagliard L (2006) Inorg Chem 45:803–807
Weck PF, Kim E, Balakrishnan N, Poineau F, Yeamans CB, Czerwinski KR (2007) Chem Phys Lett 443:82–84
Evarestov RA, Bandura AV, Losev MV, Kotomin EA, Zhukovskii YF, Bocharov D (2008) J Comput Chem 29:2079–2091
Zeng XL, Ju XH, Xu SY (2012) Adv Mater Res 550:2810–2813
Froment P, Vanbegin J, Cara J, Ronneau C (2001) J Radioanal Nucl Chem 247:285–290
Joseph B, Venkatesan KA, Nagarajan K, Srinivasan TG, Vasudeva Rao PR (2011) J Radioanal Nucl Chem 287:167–171
Tiferet E, Zalkind S, Mintz MH, Jacob I, Shamir N (2007) Surf Sci 601:936–940
Kumar A, Singhal RK, Rout S, Narayanan U, Karpe R, Ravi PM (2012) J Radioanal Nucl Chem. doi:10.1007/s10967-012-1825-8
Labroche D, Dugne O, Chatillon C (2003) J Nucl Mater 312:50–66
Long Z, Liu K, Bai B (2010) J Alloy Compd 491:252–257
Simion CA, Berinde A (2006) J Radioanal Nucl Chem 270:621–628
Fujii T, Moynier F, Agranier A, Ponzevera E, Abe M, Uehara A, Yamana H (2012) J Radioanal Nucl Chem. doi:10.1007/s10967-012-2181-4
Espenson JH (1995) Chemical kinetics and reaction mechanism. MCGraw-Hill Inc, New York
Frisch MJ, Trucks GW, Schlegel HB et al (2003) Gaussian 03 [CP]. Gaussian Inc, Pittsburgh
Shuai MB, Hu HR, Wang X, Zhao PJ, Tian AM (2011) J Mol Struc-Theochem 536:269–276
Brillant G, Gupta F, Pasturel A (2011) J Nucl Mater 412:170–176
Acknowledgments
This work was financially supported by the National Science Foundation of China (Grant No. 21101070).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Zeng, XL., Huang, SQ. & Ju, XH. Ab initio study on the reaction of uranium with oxygen. J Radioanal Nucl Chem 298, 481–484 (2013). https://doi.org/10.1007/s10967-013-2442-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-013-2442-x