
Abstract
Detection of anthropogenic noble gas isotopes in the atmosphere is an important indication that a below ground nuclear-test has taken place. Diffusion plays a critical role in the transport of these gases through the geological media to the surface where they can be detected. Better techniques are need with which to study the diffusion of noble gases through porous systems. Here we demonstrate the suitability of using prompt gamma activation analysis to measure the time dependent concentration of argon as a result of its diffusion through a porous medium that is saturated with nitrogen at atmospheric pressure. The experiments were conducted in a 1 m long tube, 10 cm diameter, and packed with fine SiO2 sand. Prompt gamma activation analysis was used to measure the concentration of argon within the experimental system as a function of time.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The presence of anthropogenic radioisotopes provides the only definitive evidence that a clandestine nuclear weapon test has taken place [1]. Noble gas isotopes are particularly useful in this regard as their non-reactive nature allows them to migrate to the surface after a below ground detonation. The International Monitoring System of the Comprehensive Nuclear-Test Ban Treaty Organization often uses the presence of anthropogenic xenon in the atmosphere [1, 2] to determine whether a below ground test has taken place. Recently, it has been suggested that anthropogenic argon could be also be used for this purpose. The detection of anthropogenic noble gas isotopes can also be useful as an indicator for underground contamination from buried radioactive materials [3]. In both cases, subsurface gas transport will affect the quantities of gas that reach the surface and the time required to do so [2, 4].
The time dependent concentration of a gas moving through a porous medium can be described using:
Here C A is the concentration (moles m−3), v is the bulk velocity (m s−1), D AB is the binary diffusion coefficient (m2 s−1) of A through B, and R A is the rate at which A is produced or lost at a given location (moles m−3 s−1) [5, 6].
All experimental techniques that are used to study the diffusion of gases through a porous medium require the ability to measure the time dependent concentration of the gas [7–13]. Common methods for doing this include gas chromatography and mass spectroscopy [8, 10, 14], photothermal deflection [15], neutron radiography [16], and nuclear magnetic resonance [17, 18]. The selection of one or another technique is based on a consideration of its availability, precision, the way the measurement needs to be conducted (invasive or non-invasive), and the type of gas under study [19]. Mass spectroscopy (sometimes combined with gas chromatography analysis) is arguably the most popular technique to analyze noble gas concentrations. The principal advantages of this technique (in a inductively coupled plasma mass spectrometer) are the small detection limits (parts per trillion), high sensitivity, and the ability to measure the elemental isotopic content of the analyte [20–22]. However, this analytical technique is invasive (requires the extraction of a sample form the system), destructive (the sample will be destroyed) and may necessitate a time-consuming sample preparation (extraction, purification and separation of the noble gas are needed previous to the analysis) [20, 21].
Prompt gamma activation analysis (PGAA) is a technique suitable for making non-invasive measurements of noble gas concentrations in porous and non-porous systems. This technique can be used to make non-invasive measurements on the concentration of noble gases as well as differentiate isotopes of the same chemical species [23, 24]. Unlike standard neutron activation analysis, PGAA is able to quantify the concentrations of non-radioactive isotopes. Here, prompt gamma photons of distinctive energy are produced in the stable isotope nucleus after it interacts with an incident neutron. These prompt gammas are detected, analyzed, and registered based on their energies using a semiconductor crystal detector in conjunction with a signal amplifier and a multichannel analyzer [25]. The gamma intensity at specific energies is proportional to the concentration of a particular non-radioactive isotope in the sample.
In this paper we demonstrate that use of PGAA for measuring time dependent argon concentrations as a result of diffusion through a homogeneous porous media.
Methodology
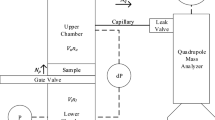
Diffusion experiments with high purity argon in nitrogen within a uniform porous media were conducted at the Nuclear Engineering Teaching Laboratory at The University of Texas at Austin. The diffusion experiments were conducted in a 1 meter long steel pipe with an internal diameter of 10.3 cm (4 in diameter ANSI Schedule 40) packed with 20/30 sieve Ottawa sand. On each end of this body, a 10 cm long by 10 cm internal diameter cylindrical aluminum chamber is added. These two chambers allow for a uniform diffusion front during the experiments. Two OMEGA® PX653-05D5 V differential pressure transducers were connected at the inlet and outlet of the aluminum chambers to guarantee measure pressure differentials and guarantee a diffusion-dominant process during the experiments. Figure 1 shows a schematic of the experimental set-up.

Experimental set-up schematic
The neutron source for this work was a 1.1 MW TRIGA Mark II nuclear research reactor. A parabolic focusing element at the end of the horizontal neuron guide allows an equivalent thermal neutron flux of 5.3 × 106 cm−2 s−1 at the sample location when the reactor operates at 950 kW [26, 27]. The 65 % relative efficiency p-type ORTEC high purity germanium detector with a resolution of 1.95 keV at 1,332 keV was used for this work and was shielded with a combination of different materials from lead bricks to a borated foam and cadmium sheets. A Tennelec TC 702 amplifier and a 16 k Canberra 8713 analog to digital convertor with a Multiport II Canberra multichannel analyzer was employed for the signal processing. Genie™ 2000 software was used to analyze the gamma photon spectrum obtained.
Calibration and detection limits
For the present study, the detection limits of argon and natural xenon were estimated. First, the total counting efficiency and energy calibration of the PGAA system were performed using a 152Eu source from the National Institute of Standards and Technology. Equation (2) shows the least square curve fit equation obtained for the counting efficiency e as a function of energy E (keV). Equation (3) gives the relationship between gamma energy and detector channel:
where:
with X denoting the detector channel.
The counting and pressure detection limits (L D(counts) and L D(pressure)) were determined to evaluate the viability to use this nuclear analysis technique to track the argon and xenon concentrations in the gas sample aluminum cylinder. The counting detection limits L D(counts) (minimum amounts of xenon and argon gases that will be detected using the current measuring process) were calculated from the critical limit, L C, following Currie’s method [28–30]. The critical limit is defined by Currie as the minimum significant value that discriminates a net signal from the background [28, 29]. Equation (4) gives the statistical calculation of the critical limit. In this expression, k is the abscissa of the standardized normal distribution corresponding to the desired error probability, and μ C is the background continuum at the channels where the peak of interest would be located. Using a false positive and false negative error probability of 5 %, the abscissa of the normal distribution, k, equals 1.624. To determine the counting detection limits, a background sample (gas sample cylinder vacuum up to 0.667 kPa (abs)) was analyzed during a 4 h live-time period with the nuclear reactor operating at 950 kW, i.e., with a 5.3 × 106 cm−2 s−1 neutron flux approximately:
The pressure and concentration detection limits, and the prompt gamma counting rates were calculated using Eqs. (6, 7, and 8) respectively, and assuming an ideal gas behavior of the sample. These equations give an estimation of the pressure detection limit L D(pressure) (kPa), the molar concentration detection limit L D(concentration) (mol cm−3) and the counting rate CR (s−1) for a detector efficiency e, a neutron flux ϕ (cm−2 s−1), a neutron-gamma isotopic cross section σγ (barns), an ideal gas constant R (8.31446 J mol−1 K−1), a reference sample pressure p (Pa) and temperature T (K), and an effective volume V eff (16 cm−3 were estimated from the geometry of the beam and sample cylinder). The net peak area or net number of counts [net countssample in Eq. (6)] were obtained from the spectra of a 99.9999 % high purity argon, and a natural xenon samples at 106.92 kPa (abs) and 298 K analyzed during a 4 h live-time period with the nuclear reactor operating at full power (950 kW). The sample spectra were evaluated using the second difference peak locator and the peak area analysis tools from Genie™ 2000. Part of the spectrum acquired for each noble gas sample is shown in Fig. 2. The estimated counting rates and the calculated detection limits are tabulated in Table 1. This table also shows the counting rates obtained for these two gases using Genie™ 2000 for comparison with their estimated values. It is important to mention that the neutron-gamma cross sections listed in Table 1 for the xenon isotopes match the values reported by the International Atomic Energy Agency and published by Molnar [31]. However, the cross sections used for the argon nuclides correspond to those listed by Molnar and reduced by a factor of 0.417. This factor was estimated from the comparison between the argon mass concentrations ρ Ar (g cm−3) calculated using the sample pressure and Eq. (9):
and the one estimated using the net peak area from the PGAA energy spectrum netcountssample, the analysis live-time t (s), and Eq. (10):

Argon spectra and counts. Top 1186.8(3) keV peak of a PGAA spectrum obtained from a pure argon sample at 106.9 kPa (abs) and 298 K. Bottom PGAA spectrum of a natural xenon sample at 107.1 kPa (abs) and 296 K
Diffusion experiments
The procedure followed for this work is divided in three major steps: porous media column venting, argon sample preparation, and diffusion of argon in nitrogen. The porous media column was vented flowing nitrogen at 25 cm3 s−1 during 1 h, and at 8.3 cm3 s−1 during at least another hour to remove the traces of argon from previous experiments. Then, the ball purge valve was closed and the experimental set-up is pressurized up to about 100 kPa.
The argon sample preparation consist of a series of vacuum-fill cycles using a Argon 6.0 Research gas from Praxair to ensure that more than 99.5 % of the gas molecules in the sample correspond to the high purity argon (Batch number W476720701). The gas sample cylinder was then pressurized up to 105 kPa (abs). Since the experimental set-up and the argon cylinder were pressurized in different rooms, the pressure of the argon sample need to be lower to match the one of the experimental set-up with a pressure excess lower than 70 Pa using a SS low-flow metering valve from Swagelok®, and a PX653-05D5V differential pressure transducer from OMEGA®. The sample was then analyzed using PGAA with the reactor operating at 950 kW.
The aluminum cylinder is connected to the experimental set-up through a full-flow quick connector from Swagelok® and the argon is allow to diffuse within the porous media for a desired amount of time. The argon concentration left within the sample cylinder was measured using PGAA and a neutron flux of about 5.3 × 106 cm−2 s−1
Error propagation
The experimental uncertainties reported in this paper were calculated following the error propagation formulae [25] and a least-squares regression analysis [32] as applied to each of the equations used to estimate the results listed in Table 1. In general, for every count net peak area used, its variance was obtained from the Genie™ 2000 peak area analysis tool after fitting a Gaussian distribution to the peak of interest in the gamma spectrum. The standard deviations for the neutron-gamma cross sections were those reported by Molnar [31] for the xenon isotopes. For the argon nuclides cross sections, the values listed in Table 1 were assumed to have an error of 10 % (close to the relative error on the values tabulated by Molnar for these isotopes). Also, the background counts and neutron flux were assumed to be the mean values and therefore equal to their variance. The error for the effective sample volume was assumed to equal 10 % (1.6 cm3). In the case of pressure and temperature, the reading accuracies (0.25 % and 0.5 °C respectively) were taken as the dominant error. The variance in the detector efficiency was determined from a least-squares regression analysis.
Results and discussion
Various diffusion experiments were conducted as described in the previous section. A gas sample cylinder was prepared and analyzed using PGAA before and after the nine diffusion experiments listed in Table 2. This table shows the live-time of prompt gamma analyses, and the percentage of argon concentration that remains within the cylinder after each test. The percentage of the remainder argon in the sample container was obtained from the ratio of the net areas of the 1186.6 keV peaks in the pre- and post-diffusion spectra. The spectrum peaks at this energy were selected despite their smaller neutron-gamma cross sections because of the higher Compton and more background peaks near-by 167.3 keV. The results recorded in this table are also depicted in Fig. 3.

Argon concentration ratio. Ratio of argon concentration within the gas sample cylinder to its initial molar concentration after diffusing through the experimental set-up for different periods of time
Conclusions
An experimental set-up, and a test procedure have been developed to demonstrate the suitability of using PGAA to evaluate the time dependent concentration of argon as a result of its diffusion through a porous media. The results demonstrate that PGAA can be used to study the time varying concentrations of argon and xenon that would result from their diffusion through a porous medium.
References
Auer M, Kumberg T, Sartorius H, Wernsperger B, Schlosser C (2010) Ten years of development of equipment for measurement of atmospheric radioactive xenon for the verification of the CTBT. Pure Appl Geophys 167(4):471–486
Zähringer M, Becker A, Nikkinen M, Saey P, Wotawa G (2009) CTBT radioxenon monitoring for verification: today’s challenges. J Radioanal Nucl Chem 282(3):737–742
Dresel P, Olsen K, Hayes J, McIntyre J, Waichler S, Kennedy B (2008) Environmental applications of stable xenon and radioxenon monitoring. J Radioanal Nucl Chem 276(3):763–769
Wotawa G, Becker A, Kalinowski M, Saey P, Tuma M, Zähringer M (2010) Computation and analysis of the global distribution of the radioxenon isotope 133Xe based on emissions from nuclear power plants and radioisotope production facilities and its relevance for the verification of the nuclear-test-ban treaty. Pure Appl Geophys 167(4):541–557
Bird RB, Stewart WE, Lightfoot EN (2007) Transport phenomena, 2nd edn. Willey, New York
Deen WM (1998) Analysis of transport phenomena. Topics in chemical engineering. Oxford University Press, Inc., New York
GarcÌa-GutiÈrrez M, Mingarro M, Missana T, MartÌn PL, Sedano LA, Cormenzana JL (2004) Diffusion experiments with compacted powder/pellets clay mixtures. Appl Clay Sci 26(1–4):57–64
Allaire SE, Lafond JA, Cabral AR, Lange SF (2008) Measurement of gas diffusion through soils: comparison of laboratory methods. J Environ Monit 10(11):1326–1336
Abu-El-Sha’r W, Abriola LM (1997) Experimental assessment of gas transport mechanisms in natural porous media: parameter evaluation. Water Resour Res 33(4):505–516
Rees KCJV, Sudicky EA, Rao PSC, Reddy R (1991) Evaluation of laboratory techniques for measuring diffusion coefficients in sediments. Environ Sci Technol 25(9):1605–1611
Takeda M, Nakajima H, Zhang M, Hiratsuka T (2008) Laboratory longitudinal diffusion tests: 1. Dimensionless formulations and validity of simplified solutions. J Contam Hydrol 97(3–4):117–134
Vaatainen K, Timonen J, Hautojarvi A (1993) Development of a gas method for migration studies in fractured and porous media. In: Proceedings of the 16th international symposium on the scientific basis for nuclear waste management (November 30, 1992–December 4, 1992, Boston. Materials research society symposium proceedings). Materials Research Society, pp 851–856
Currie JA (1960) Gaseous diffusion in porous media Part 1. A non-steady state method. Br J Appl Phys 11(8):314
Islas-Juarez R, Samanego-V F, Luna E, Perez-Rosales C, Cruz J Experimental study of effective diffusion in porous media. In: 2004 SPE international petroleum conference in Mexico (November 7, 2004–November 9, 2004, Puebla, Mexico). Society of Petroleum Engineers (SPE), London, pp 781–787
Rohling JH, Shen J, Wang C, Zhou J, Gu CE (2007) Determination of binary diffusion coefficients of gases using photothermal deflection technique. Appl Phys B 87(2):355–362
Takai S, Mandai T, Kawabata Y, Esaka T (2005) Diffusion coefficient measurements of La2/3–xLi3x TiO3 using neutron radiography. Solid State Ionics 176(29–30):2227–2233
Mair RW, Wong GP, Hoffmann D, Hurlimann MD, Patz S, Schwartz LM, Walsworth RL (1999) Probing porous media with gas diffusion NMR. Phys Rev Lett 83(16):3324
Mair RW, Rosen MS, Wang R, Cory DG, Walsworth RL (2002) Diffusion NMR methods applied to xenon gas for materials study. Magn Reson Chem 40:S29–S39
Pereira LM, Souza RD, Orlande HRB, Cotta RM (2001) A comparison of concentration measurement techniques for the estimation of the apparent mass diffusion coefficient. Braz J Chem Eng 18:253–265
Beyerle U, Aeschbach-Hertig W, Imboden DM, Baur H, Graf T, Kipfer R (2000) A mass spectrometric system for the analysis of noble gases and tritium from water samples. Environ Sci Technol 34(10):2042–2050
Mark DF, Stuart FM, de Podesta M (2011) New high-precision measurements of the isotopic composition of atmospheric argon. Geochim Cosmochim Acta 75(23):7494–7501
Technologies A (2001) Inductively coupled plasma mass spectrometry. Course Number H8974A. Agilent Technologies
Revay Z (2008) Prompt gamma activation analysis of samples in thick containers. J Radioanal Nucl Chem 276(3):825–830
Rios Perez C, Lowrey J, Biegalski S, Deinert M (2012) Xenon diffusion studies with prompt gamma activation analysis. J Radioanal Nucl Chem 291(1):261–265
Knoll G (2010) Radiation detection and measurement, 4th edn. Wiley, Hoboken
Harrison RK, Landsberger S (2009) Determination of boron over a large dynamic range by prompt-gamma activation analysis. Nucl Instrum Methods Phys Res Sect B 267(3):513–518
Rèvay Z, Harrison RK, Alvarez E, Biegalski SR, Landsberger S (2007) Construction and characterization of the redesigned PGAA facility at The University of Texas at Austin. Nucl Instrum Methods Phys Res Sect A 577(3):611–618
Currie LA (1968) Limits for qualitative detection and quantitative determination. Application to radiochemistry. Anal Chem 40(3):586–593
Currie LA (1995) Nomenclature in evaluation of analytical methods including detection and quantification capabilities. Pure Appl Chem 67(10):25
Biegalski SR, Green TC, Alvarez E, Aghara S (2007) Background characterization of The University of Texas PGAA facility. J Radioanal Nucl Chem 271(2):413–417
Molnar GL (ed) (2004) Handbook of prompt gamma activation analysis with neutron beams. Kluwer Academic Publishers, Dordrecht
Figliola RS, Beasley DE (2000) Theory and design for mechanical measurements, 3rd edn. Wiley, New York
Acknowledgments
The authors would like to thank Dr. Carlos Hidrovo for discussions about diffusion of gases through porous media, and the staff of the Nuclear Engineering Teaching Laboratory for their help with the prompt gamma measurements. This material is based upon work supported by the Department of Energy, National Nuclear Security Administration under Award Number DE-AC52-09NA28608.
Disclaimer
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government, nor any agency thereof, nor any of their employees, makes any warranty, expressed or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would infringe privately owned rights. Reference herein to any specific commercial product, process, or service by name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation or favoring buy the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rios Perez, C.A., Biegalski, S. & Deinert, M.R. Measuring the diffusion of noble gases through a porous medium using prompt gamma activation analysis. J Radioanal Nucl Chem 296, 951–956 (2013). https://doi.org/10.1007/s10967-012-2003-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-012-2003-8