
Abstract
Ion-exchange resins and activated charcoal beds are employed for purification of the cooling water that is pumped through the core of pool type nuclear research reactors. Once expended, these media are replaced and become radioactive wastes that contain low concentrations of long-lived fission and activation products, uranium isotopes and transuranium elements. Determination of the radioactive inventory is of paramount importance in the management of such radioactive wastes, which, besides high-energy photon emitters that can be identified and quantified directly by gamma-ray spectrometry, also contain pure alpha, pure beta and low-energy photon emitters whose quantitative determination require radiochemical separation. These later are collectively known as difficult to measure (DTM) radionuclides. A characterization program embracing the DTM radionuclides is currently in progress for spent ion-exchange resins and activated charcoal beds that were definitively withdrawn from the water cleanup system of the IEA-R1 nuclear research reactor. Radiochemical methods used in the characterization program include separations with specific anionic resins, chromatographic extractions and co-precipitation, which enabled the measurement of the activity concentrations of 90Sr, 234U, 235U, 238U, 238Pu, 239+240Pu, 241Pu, 241Am and 244Cm. An enhanced retention of uranium and transuranium elements was observed in the activated charcoal compared to the ion-exchange resins as a result of the tendency of actinides to undergo hydrolysis in aqueous solutions.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The IEA-R1 is a 5 MW pool type nuclear research reactor, moderated and cooled by light water, located at the Institute for Nuclear and Energy Research in São Paulo (IPEN/CNEN-SP) and used for scientific research as well as for production of radioisotopes [1, 2]. Cooling water is pumped through the reactor core to remove the heat generated by nuclear reactions. Impurities in the cooling water may become radioactive as a result of interaction with radiations in the reactor and must, therefore, be continuously removed from the coolant to avoid scale up of radioactivity in the system. High purity cooling water is obtained using a cleanup system consisting of cartridge filters, activated charcoal beds and ion-exchange resin beds, which retain radionuclides by filtration, adsorption and ion exchange.
The water cleanup system of the IEA-R1 research reactor has six parallel polypropylene cartridge filters, two parallel mixed ion-exchange resin beds and two parallel activated charcoal beds. Once a year, the mixed ion-exchange resin beds are regenerated with concentrated sulfuric acid and concentrated sodium hydroxide, and the activated charcoal beds are back flushed with deionized water.
When these media are saturated and no more able to maintain the electrical conductivity of the cooling water below the design limits, they are replaced and become low-level radioactive waste. They contain activation products generated by irradiation of structural materials in the reactor core, along with fission products and isotopes of uranium and transuranium elements that escaped from the irradiated nuclear fuel or have been generated by irradiation of the slight surface contamination of nuclear fuel elements [3].
The management of such radioactive wastes demands the determination of their radioactive inventory as a first and fundamental step. The activity concentration of some typical radionuclides contained in radioactive wastes can be measured directly by gamma-ray spectrometry of samples or gamma-ray scanning of the complete waste package, for which is necessary a previous energy and efficiency calibration of the measuring apparatus, a rigorous control of its geometry and an accurate evaluation of all attenuation effects involved.
However, many other radionuclides commonly found in radioactive wastes are pure alpha, pure beta or low-energy photon emitters not amenable to direct measurement. This is the reason why they are designated as difficult to measure (DTM) radionuclides [4] and require the use of radiochemical methods on waste samples before their activity concentrations can be determined. The present work describes the determination of some DTM radionuclides of the elements strontium, uranium, neptunium, plutonium, americium and curium from ion-exchange resins and activated charcoal beds that were withdrawn from the water cleanup system of the IEA-R1 nuclear research reactor.
Experimental
Materials and equipment
Analytical grade reagents and certified calibrated radiation sources were used in the analysis performed in the scope of the radiochemical methods. All the standards were supplied by the US National Institute of Standards and Technology, NIST.
UTEVA® resin and TRU resin used for chromatographic extractions were purchased as pre-packed columns. SR resin was packed in 80 mm length and 8 mm diameter glass columns. All these resins were supplied by Eichrom Technologies, Inc.
Strongly anionic ion-exchange resin (Dowex® 1X8 chloride form, 50–100 mesh, from Sigma-Aldrich Inc., USA) was packed in 200 mm length and 8 mm diameter glass columns.
The measurement of alpha-emitters present in the samples was performed with an Alpha Analyst Integrated Alpha Spectrometer from Canberra Industries. The system is provided with passivated implanted planar silicon detectors with 450 mm2 active detection area, 18 keV (FWHM) resolution, counting efficiency of 17–19 % for a source–detector distance of 5 mm and calibrated for energies ranging from 3 to 10 MeV. A standard source certified by Eckert and Ziegles Analytics with electrodeposited 234U, 238U, 239Pu and 241Am was used to calibrate the system. Genie™ 2000 software, from Canberra Industries, was used for data acquisition and analysis.
Beta-emitters were measured with an automatic HIDEX model 300SL liquid scintillation counter, using triple to double coincidence ratio to automatically correct quenching with an energy discriminator to separate alpha and beta for simultaneous counting. MikroWin Hidex 2000 software was used for data acquisition and processing.
The measurements of 60Co and 137Cs activity concentrations were performed by gamma-ray spectrometry, employing a P-type HPGe detector from Canberra Industries USA, which has a volume of 192.5 cm3 and a 0.5 mm-thick beryllium window, with 1.9 keV (FWHM) resolution and 45 % relative efficiency for the 1,332.5 keV gamma-ray of 60Co.
Sample preparation
Multiple samples were taken from some drums to check the homogeneity of the waste when different phases were visible within it.
Forty-two samples were taken from 14 200 L-drums containing activated charcoal and 36 samples were taken from seven 200 L-drums containing ion-exchange resins stored in the Radioactive Waste Management Department of IPEN/CNEN-SP. Each sample was analyzed for 60Co and 137Cs by gamma-ray spectrometry, and for Sr, U, Np, Pu, Am and Cm isotopes by radiochemical methods.
Ion-exchange resin and activated charcoal samples of approximately 5 g were dried in an oven at 60 °C for 24 h. Aliquots of 2 g from each dried sample were then prepared in a sealed vial with calibrated geometry for gamma-ray spectrometry to determine the activity concentrations of 60Co and 137Cs. Approximately 1.5 g aliquots of each sample were taken for total dissolution and further determination of DTM radionuclides activities.
Dissolution of ion-exchange resin and activated charcoal samples
In order to dissolve the resins, 1.5 g aliquots of each sample were digested in Teflon beakers on a hot plate with 3 × 10 mL of concentrated nitric acid, 5 mL of 30 % hydrogen peroxide and 10 mL of concentrated perchloric acid. The sample solution was evaporated to dryness and the residue was dissolved with 10 mL of 8 mol L−1 HNO3.
For activated charcoal samples, approximately 1.5 g aliquots of each sample were calcined at 450 °C in oven with a programmable heating rate for 24 h. The residue was digested in Teflon beaker on a hot plate with 3 × 10 mL of concentrated nitric acid, 5 mL of concentrate hydrofluoric acid, 5 mL 30 % hydrogen peroxide and 10 mL concentrated perchloric acid. Then, the solution was evaporated and the remaining salt residue dissolved with 10 mL of 8 mol L−1 HNO3.
Previously dissolved ion-exchange resin and charcoal samples were diluted with 8 mol L−1 HNO3 to 100 mL in order to form the stock solutions used in subsequent determinations.
Separation of Pu and Np
Samples for isotope sequential analysis from each stock solution were spiked with 0.05 Bq of 232U, 236Pu/242Pu, 243Am tracers and 1 mL of stable strontium carrier solution (0.1 g L−1) was added. The oxidation state of Pu and Np was then adjusted with ferrous sulphamate (0.1 mL iron nitrate 5 mg mL−1 + 0.5 mL 1.5 mol L−1 sulfamic acid) and ascorbic acid (1.25 mL of 1.5 mol L−1), followed by the addition of 2–3 g of sodium nitrite [5].
Separation of Pu and Np from other radionuclides was carried out with Dowex® 1X8 chloride form resin as follows: a 50 mL sample were loaded through an anionic column previously conditioned with 8 mol L−1 HNO3 and the column was rinsed with 30 mL of 8 mol L−1 HNO3. Under these conditions, only Pu4+ and Np4+ were retained in the column, and the rinsed solution were collected to recover Sr, U, Am and Cm isotopes. The column was washed with 9 mol L−1 HCl to remove interferences and to change the medium from nitrate to chloride. Retained Pu4+ and Np4+ were selectively eluted by reduction of their oxidation state with 0.5 g of hydroxylamine chlorhydrate placed on the top of the column followed by 30 mL 0.5 mol L−1 HCl. This eluted fraction was evaporated and the residue dissolved with 1 mol L−1 HNO3 and diluted with deionized water to 25 mL. An aliquot was taken for the determination of 241Pu [6, 7] by liquid scintillation counting and the remaining volume was used for the electrodeposition on silver planchets in 0.8 mol L−1 ammonium sulfate with the pH adjusted to 2–3 for 90 min, applying a current of 1.2 A [8]. Once the electrodeposition process has been completed, Pu and Np were quantified by alpha spectrometry [8].
The aliquot for 241Pu determination was evaporated in a counting vial for liquid scintillation and the precipitated salts were dissolved with 1 mL of 0.1 mol L−1 HNO3. Scintillation cocktail Ultima-Gold AB™ from Perkin Elmer was added and then 241Pu was counted for 300 min [6, 9].
Separation of Sr, U, Am and Cm
Strontium isotopes, U, Am and Cm, present in the effluent from the anionic resin used in the first step of the method, were purified with UTEVA® chromatographic resin specific for U isotopes, TRU-spec resin for Am and Cm isotopes and SR-spec resin specific for Sr isotopes [5, 10–12]. The effluent was evaporated and the residue dissolved in 3 mol L−1 HNO3. The columns were mounted in tandem for percolation of samples, washed with 3 mol L−1 HNO3 and disconnected in the elution process [13, 14].
Uranium retained in the UTEVA® column was eluted with 15 mL 0.1 mol L−1 HCl, americium and curium retained in the TRU-spec column were eluted with 15 mL 0.05 mol L−1 HNO3 and strontium retained in the SR-spec column was eluted with 15 mL 0.05 mol L−1 HNO3.
The eluted fractions of Am and Cm were electrodeposited on silver planchets, in 0.8 mol L−1 ammonium sulfate with the pH adjusted to 2–3 for 90 min, applying a current of 1.2 A [8]. These radionuclides were then quantified by alpha spectrometry.
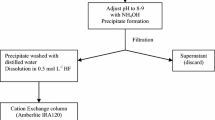
The last effluent in the sequential separation, which contains the Sr isotopes (approximately 30 mL), was co-precipitated with 0.3 g of oxalic acid and the pH was adjusted to 9–10 with concentrated ammonium hydroxide. The precipitate was filtered through polypropylene filters with 0.1 μm and 25 mm diameter (Pall Corporation, USA). Chemical recovery of the precipitate was gravimetrically determined. The precipitate was dissolved with 1 mL 0.1 mol L−1 HCl in the liquid scintillation counting vial, the Ultima-Gold AB™ scintillation cocktail was added and then 90Sr was counted [11, 15]. Figure 1 shows the steps for sequential determination of Pu, Np, U, Am, Cm and Sr isotopes.

Flow diagram of the radiochemical sequential determination of Sr, U, Np, Pu, Am and Cm isotopes
Results and discussion
Sample preparation
The use of anionic resin in the first step of the separation process has the following advantages when compared to the direct chromatographic resin separation process: (a) larger sample volumes can be loaded; (b) some interfering elements present in the analyzed matrix can be eliminated; (c) costs are reduced because the columns can be regenerated.
Dissolution of ion-exchange resin and activated charcoal samples
The complete dissolution of ion-exchange resins is a difficult task because these materials are designed to withstand the attack of strong acids, alkalis and solvents. Digestion of the samples by microwave is a faster alternative, but the total sample mass in the sample holder is limited to 0.1 g, which is insufficient for the determination of many radionuclides.
Radionuclides separation
Chemical recovery of the analytes was obtained using tracers and carriers added in the sample dissolution process. Standard solutions of 232U, 236Pu/242Pu and 243Am and strontium carbonate carrier were used to determine chemical recovery of the corresponding elements. The 236Pu standard was used in the determination of 237Np because the alpha energy emitted by 242Pu is close to the alpha emission energy of 237Np.
Anionic exchange chromatography was applied in the separation and purification of Sr, U, Np, Pu, Am and Cm. This is one of the most selective techniques employed for radiochemical separation of actinides, since elements with high valence states tend to form anionic complexes easily [16].
The affinity and the distribution coefficient between ion-exchange resins, solvent or chromatographic resin used in the separation process depends on the oxidation state of the elements. Those which form nitrate complexes and are retained in anionic exchangers from concentrated nitric solutions are: Th(IV), Pa(IV), Np(IV), Pu(IV), Pd(IV), Pd(II), Au(III), Re(VII) and Tc(VII). These metals are easily separated from chemical species that are not adsorbed, such as Al(III), Fe(II and III), alkali and alkaline earth metals, rare earths elements and trivalent actinides [17].
Pu(IV) and Np(IV) also form chloride complexes and can be separated from Th(IV) which does not form complexes when the medium is changed from nitrate to chloride: both stay in the column while Th(IV) is eluted. Usually, the loading solution is in an 8 mol L−1 nitric medium and Th is removed by loading 9–10 mol L−1 HCl. Pu and Np were eluted with diluted HCl in the presence of a reducing agent. Sr, U, Am and Cm are not retained in anionic columns; the effluent is used for sequential separation of these elements [18, 19].
Due to the prevailing unavailability of 241Pu certified standards, the efficiency calibration of the liquid scintillation counter was performed using a 3H standard [9, 12].
The choice of silver planchets for alpha-emitters electrodeposition is based on the thin and uniform sources thus obtained, which result in good alpha spectra resolution and good measurement reproducibility.
Chemical recovery of the analytical methods were 62–85 % for Sr, 75–99 % for U, 60–95 % for Np/Pu and 45–85 % for Am/Cm. The activity concentrations of 237Np in all samples were below the 0.002 Bq g−1 detection limits for 1 g aliquots of activated charcoal and ion-exchange resins. The concentrations of 239Pu and 240Pu are expressed together because the energies of their alpha emissions are so close [20] that can not be differentiated by alpha spectrometry [21].
The detection limit for gamma-emitters was 0.030 Bq g−1 for 1.5 g aliquots and measurement of 3,600 s of live time.
Statistical data
The results of activity concentration of each radionuclide were normalized to the date of discharge from the water cleanup system of the IEA-R1 nuclear research reactor. The medians of the results, along with the interquartile ranges (IQR), minima and maxima, are presented in Table 1.
Figure 2 shows the box plot graph of the results, after possible outliers were excluded from the data sets. These statistics were chosen, instead of the mean and the standard deviation, because they are less sensitive to the extreme values observed, which span many orders of magnitude in the case of some of the radionuclides analyzed. The IQR, used in the calculations for exclusion of outliers, is the difference between the 25 and 75 % percentiles of concentration in each data set.

Activity concentration of each radionuclide measured in the spent beds of activated charcoal and ion-exchange resin
There are many probable outliers signaled in Fig. 2. Concerning 60Co in activated charcoal samples, the considered outliers could probably be attributed to a process of partial desorption of cobalt during the annual back flush of the charcoal bed while the material was still in the cleanup system, and to the inhomogeneous distribution of the material when packaged in the waste drums.
The outliers observed in ion-exchange resin samples are certainly the result of some degree of segregation of the anionic and the cationic fractions of the resin while the mixed bed was being transferred to the waste drums. In the case of transuranium elements, the outliers are a consequence of the large uncertainties in the measurements due to the very low concentrations in the ion-exchange resin.
It is evident from Fig. 2 that the heavier elements are preferentially retained in the activated charcoal bed, which is positioned before the ion-exchange resin bed in the flow direction of the coolant. This can be attributed to the propensity of metallic cations to undergo hydrolysis in aqueous solutions. The more charged the metallic cation, the stronger the interaction with water molecules is. Actinides such as U, Pu, Am and Cm have a tendency to form polymers of colloidal dimensions, with molecular mass as high as 1010 atomic mass units. The actinyl ions MO2 + and MO2 2+ are less acidic in their nature than M4+ ions, and therefore have less of a tendency to undergo hydrolysis, a property that decreases in the following order: M4+ > MO2 2+ > M3+ > MO2 + [11, 16, 17]. The consequence of this property is observed in Fig. 2, where the isotopes of uranium and transuranium elements are more concentrated in the activated charcoal beds than in the ion-exchange resin beds, as a result of the more prevailing process of surface adsorption than ion exchange.
Conclusion
A radiochemical sequential separation method was developed in this work to determine the radionuclides 90Sr, 234U, 235U, 238U, 237Np, 238Pu, 239+240Pu, 241Pu, 241Am and 244Cm in samples of activated charcoal and ion-exchange resin. This method was effective at characterizing DTM radionuclides in radioactive wastes. The use of anionic ion-exchange and chromatographic resins in the separation process is efficient because of the high chemical recovery of the tracers and carriers employed. The use of anionic resin in the first step of the separation process allows the elimination of interfering radionuclides like 60Co, 137Cs and 55Fe which otherwise would hinder the determination of the beta-emitters of interest.
References
Vasconcellos MBA, Saiki M (2006) Radiochemistry teaching and research activities in Brazil. J Radioanal Nucl Chem 270:263–267
Vasconcellos MBA, Saiki M, Fávaro DIT, Maihara VA, Figueiredo AMG, Catharino MGM (2004) Neutron activation analysis at the research reactor center of IPEN/CNEN-SP—biological and environmental applications. J Radioanal Nucl Chem 259:489–492
IAEA, International Atomic Energy Agency (1994) Classification of radioactive waste. Safety Series 111-G-1.1. Vienna
IAEA, International Atomic Energy Agency (2009) Determination and use of scaling factors for waste characterization in nuclear power plants. Nuclear Energy Series No. NW-T-1.18. Vienna
Eichrom methods, americium, neptunium, plutonium, thorium, curium, uranium, and strontium in water (with vacuum box system) (2006) Analytical procedures ACW 17 VBS Rev. 1.0. Eichrom Technologies, Inc., Lisle, pp 1–17
Payne RF, Clark SB, Elliston JT (2008) Radioanalytical approach to determine 238Pu, 239+240Pu, 241Pu and 241Am in soils. J Radioanal Nucl Chem 277:269–274
Paatero BJ, Jaakkola T (1994) Determination of the 241Pu deposition in Finland after the Chernobyl accident. Radiochim Acta 64:139–144
Talvitie NA (1972) Electrodeposition of actinides for alpha spectrometric determination. Anal Chem 44:280–283
Ikäheimonen TK (2000) Measurement of 241Pu in environmental samples. J Radioanal Nucl Chem 243:535–541
Rodríguez M, Gascón JL, Suárez JA (1997) Study of interferences in the determination of Pu, Am and Cm in radioactive waste by extraction chromatography. Talanta 45:181–187
Tavcar P, Smodis B, Benedik L (2006) Radiological characterization of low- and intermediate-level radioactive wastes. J Radioanal Nucl Chem 273:593–596
Horwitz EP, Chiarizia R, Dietz ML, Diamond H, Nelson D (1993) Separation and pre-concentration of actinides by extraction chromatography using a supported liquid anion exchanger: application to the characterization of high-level nuclear waste solutions. Anal Chim Acta 281:361–372
Maxwell SL III, Culligan BK (2006) Rapid column extraction method for actinides in soil. J Radioanal Nucl Chem 270:699–704
Maxwell SL III, Faison DM (2007) Rapid column extraction method for actinides and strontium in fish and other animal tissue samples. J Radioanal Nucl Chem 275:605–612
RADCHEM (2006) Radiochemical procedures for the determination of Sr, U, Pu, Am and Cm. Institute for Energy Technology, Norway. ISBN 87-7893-185-1, NKS-124
Korkisch J (1989) Handbook of ion-exchange resins: their applicability to inorganic analytical chemistry, vols I and II. CRC Press Inc., Boca Raton
Seaborg GT, Loveland WD (1990) The elements beyond uranium. Wiley, New York
Bagnall KW (1972) The actinide elements. In: Robinson PL (ed) Topics in inorganic and general chemistry. Monograph 15. Elsevier Publishing Co, Amsterdam
American Society for Testing and Materials (ASTM) (2010) Standard guide for determination of plutonium and neptunium in uranium hexafluoride by alpha spectrometry. ASTM C1561-10
Firestone RB, Shirley VS (eds) (1996) Table of isotopes, 8th edn. Wiley, New York
Spry N, Parry S, Jerome S (1999) The development of a sequential method for the determination of actinides and 90Sr in power station effluent using extraction chromatography. Appl Radiat Isot 53:163–171
Acknowledgments
The authors gratefully acknowledge support from the staff of the Radioactive Waste Management Department of IPEN/CNEN-SP in sampling the wastes.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Taddei, M.H.T., Vicente, R., Marumo, J.T. et al. Determination of long-lived radionuclides in radioactive wastes from the IEA-R1 nuclear research reactor. J Radioanal Nucl Chem 295, 951–957 (2013). https://doi.org/10.1007/s10967-012-1865-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-012-1865-0