
Abstract
Chemical separation methods in conjunction with instrumental neutron activation analysis (INAA) were developed for measuring iodine levels in commercially available bovine milk with varying milk fat (MF) content. Samples of homogenized (3.25 % MF), partly skimmed (2 % MF), partly skimmed (1 % MF), partly skimmed calcium enriched (1 % MF + Ca), and skim (<0.05 %) milk were purchased from local supermarkets. Ion exchange chromatography, solvent extraction, and ammonium sulfate precipitation methods were applied to the separation of the inorganic, lipidic and proteic fractions of iodine in milk. The levels of iodine were measured by INAA in total reactor and epi-cadmium (EINAA) neutron flux in conjunction with conventional gamma-ray and Compton suppression spectrometry (CSS). A pseudo-cyclic INAA method coupled with CSS (PC-INAA-CSS) was also explored as an instrumental option to further lower the detection limit of iodine. The detection limits of 0.06, 0.06 and 0.02 μg mL−1 for iodine were obtained using INAA-CSS, EINAA-CSS, and PC-INAA-CSS methods, respectively. Although the PC-INAA-CSS method provided the lowest detection limit, the INAA-CSS method was sufficient for the determination of total iodine in almost all samples analyzed in this work. The total iodine concentrations (μg mL−1) were: 0.40 ± 0.01 (in 3.25 % MF), 0.40 ± 0.01 (2 % MF), 0.42 ± 0.01 (1 % MF), 0.42 ± 0.01 (<0.05 %), and 0.96 ± 0.01 (1 % MF + Ca) milk samples. Iodine bound to various fractions of the milk samples analyzed, in percent of total iodine content, ranged: (0.05–1.8), (1.9–4.8), (90–95) for the lipidic, proteic and anionic inorganic fractions respectively. Iodine recovery in all cases was higher than 96 %.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Canada is one of the countries with fairly high incidence of osteoporosis. Statistics shows that 1 in 4 women and at least 1 in 8 men over 50 have this disease [1]. Perhaps this is one of the reasons for the high consumption of bovine milk and its products in Canada. In 2009, the per capita consumption of bovine milk with different milk fat (MF) content of Canadians was: 11.14 L of homogenized (3.25 % MF), 37.33 L of partly skimmed (2 % MF), 18.10 L of partly skimmed (1 % MF), 8.60 L of skimmed (<0.5 % MF), 5.73 L of chocolate, and 0.37 L of buttermilk [2]. Although the annual per capita consumption has decreased from 1998 to 2009, Canada still is among the top 10 consumers of milk and its products in the world [3].
At the same time, the iodine content of milk has been increasing worldwide, at least since the 1980s. A recent study conducted in Canada of 501 farms covering all provinces showed that the iodine content of Canadian retail milk was on the average 304 ± 8.4 μg kg−1 which is considered a high value [4]. It was also found that the Central and Atlantic Canada had higher milk iodine content compared to the Prairies and Western Canada. This work [4] encouraged to take actions to reduce iodine levels in Canadian milk. Given the facts that in Canada the table salt is iodized and that milk and milk products are consumed extensively as well, this country unlike many others in the world is not at risk of iodine deficiency disorder but instead could be at risk of high iodine intake [4, 5]. It has been reported since mid-1980s that Canadians exceed the recommended nutrient intake (RNI) value for iodine by at least 6 times [6]. High levels of iodine in a diet may inhibit the function of the thyroid gland and produce symptoms of iodine deficiency as well.
Toxicological effects of chemical elements depend on not only the concentration but also their chemical species. Total iodine content of milk has been routinely measured at least since 1970; however this is not the case for iodine speciation analysis. The small number of reports available on iodine speciation is perhaps due to the low iodine content involved and/or the volatility of this element leading to losses during the separation as well as analytical detection by most techniques. Much of the work on iodine species in milk reported so far involves iodide and iodate [7–9], T3, other organic or inorganic species [10, 11], and iodine bound to casein, whey, and/or fat [12, 13]. The commonly used analytical techniques are ICP-MS and NAA [7–13]. Both are very sensitive techniques; however, NAA does not require sample to be in the liquid phase and it is able to detect iodine instrumentally at very low levels in small sample sizes.
In 2008 a method to determine iodine and some of its species in homogenized milk (3.25 % MF) was reported by us. Here milk was separated into lipids, protein and aqueos fractions by chemical methods and their iodine levels were measured by EINAA [6]. The main advantages of this fractionation scheme include a high recovery, possibility of further fractionation, and its application to any element. In this scheme lipid and protein fractions were separated first followed by the determination of inorganic iodine in the remaining solution. Although iodine levels were successfully measured, the solution also contained high levels of Na, Mg, K, Cl, Br and other elements which caused major interferences in the iodine determination by NAA. The use of epithermal neutron irradiation in conjunction with Compton suppression counting for the 128I nuclide reduced some interference of the above mentioned elements. It is still possible that the levels of some inorganic iodine species were too low to be detected by this method. For this reason a new separation scheme for iodine fractionation in bovine milk with different MF content and NAA methods with lower detection limits are reported here. INAA in total reactor and epi-cadmium (EINAA) neutron flux in conjunction with conventional gamma-ray and Compton suppression spectrometry (CSS) has been developed. In addition, a pseudo-cyclic INAA method coupled to CSS (PC-INAA-CSS) has been developed to further lower the detection limit of iodine. The details of the separation as well as NAA methods are given below.
Experimental
Neutron activation analysis methods
Milk samples, comparator standards, and reference materials
Five types of commercially available milk sample with different MF content were purchased from supermarkets in Halifax. The samples were: homogenized (3.25 % MF), partly skimmed (2 % MF), partly skimmed (1 % MF), partly skimmed calcium enriched (1 % MF + Ca), and skim (<0.05 %) milk.
A stock solution of 4 μg mL−1 of iodine using ammonium iodide (Spex, ultrapure) was prepared to be used as the iodine comparator standard. A secondary stock solution of 1.5 μg mL−1 was prepared from the above solution. Different volumes of this solution were pipetted out to cover a mass range of 0.2–1.0 μg. Distilled deionized water (DDW) was used for making up the volume to 0.75 mL.
The NAA methods were validated using two materials, namely the NIST Non-Fat Milk Powder SRM-1549 and NIST Wheat Gluten RM-8418.
Irradiation and counting systems
Samples and standards were irradiated at the inner and the cadmium-shielded outer pneumatic sites of the Dalhousie University SLOWPOKE-2 Reactor (DUSR) facility at a neutron flux of 2.5 × 1011 cm−2 s−1. The details of the stability, homogeneity and reproducibility of the DUSR facility has previously been described [14–16]. Two detection systems were mainly used in this work. One was a Compton suppression gamma-ray spectrometry system consisting of an EG&G Ortec HPGe p-type coaxial detector with a resolution (FWHM) of 1.72 keV at the 1332.5 keV photopeak of 60Co and a relative efficiency of 25 % with respect to a standard NaI(Tl) detector in conjunction with an Ortec D-SPEC plus pulse height analyzer. The guard detector used in this system was a 10″×10″ NaI(Tl) annulus with 5 photomultiplier tubes (PMTs) supplied by Harshaw and a 3″×3″ NaI(Tl) plug with one PMT supplied by Teledyne. The peak-to-Compton ratio of this system was 582:1 at the 662-keV photopeak of 137Cs. The second counting system consisted of a D-SPEC plus multi-channel analyzer in conjunction with a Canberra 60 cm3 Ge(Li) semiconductor detector with a resolution of 1.88 keV at the 1332.5-keV photopeak of 60Co, a peak-to-Compton ratio of 35:1 and an efficiency of 9.5 %. All measurements performed on the first counting system were done at a distance of 2.6 cm in the anticoincidence mode for Compton suppression while in the second system the measurements were done on top of the detector (covered with a plastic dish) in a conventional counting mode.
Separation and determination of milk fractions
Lipid separation and determination by solvent extraction
About 25 mL of milk were placed in a separatory funnel to which 2 mL of concentrated ammonia solution were added. After shaking for 1 min, 15 mL of hexane and 25 mL of isopropanol were added, and the sample was mixed for 2 more min. Then 10 mL of hexane were added and mixed for 1 min. After the separation of phases (no centrifugation was required), the upper phase (hexane) was removed and stored. The lower phase was taken through a second extraction similar to the first one except using half of the initial volumes. After separating the upper phase in the second extraction, 10 mL of hexane were added to the bottom phase and a third extraction was carried out. The three hexane phases from each extraction were combined and washed twice with 10 mL of DDW. The organic solvent was evaporated under air in fume hood until reached constant weight.
Protein determination by bradford method
Different volumes of a standard solution of 1 mg Casein mL−1 were taken to cover a range of 4–100 μg of protein. The total volume was made up to 1 mL with water. 3 mL of Coomassie brilliant blue G-25 reagent were added, and the solution was shaken in a vortex mixer. Absorbance at 596 nm was measured immediately and without any heating. Three replicates were used to generate each point on the calibration curve. Milk samples were diluted 100 times before 100 μL were taken to carry out the assay.
Protein separation by ammonium sulfate precipitation
A saturated ammonium sulfate solution (4.06 mol L-1 at 20 °C) was added to the sample assuring that the dilution due to the sample volume makes a final salt concentration of 3 mol L−1 in the mixture. Once proteins were precipitated, a washing step using methanol followed by ketone and ethyl ether was employed. The precipitated protein was dried until constant weight.
Batch separation of inorganic iodine anionic species by ion-exchange chromatography
Cleaning of resin
About 25 g of Dowex 1X8 (100–200) mesh resin in chloride form were soaked in 5 % sodium hypochlorite solution at pH 1–2. The resin was stirred for about 30 min followed by washing with DDW until no more chloride ions were detected by the silver nitrate test. The resin was soaked in 25 % (v/v) ammonia solution and stirred overnight, washed with water until neutral pH was attained, and dried in air. A portion of the dried resin was irradiated for 1 min in the inner position of the DUSR facility, allowed to decay for 1 min, and counted for 5 min. If the 443-keV photopeak of 128I was detected in the spectrum then the cleaning procedure was repeated again until there was no iodine peak. In general, three washings were required to make the resin iodine-free and to attain a constant ratio of chlorine counts/mass. At the end, about 25 g of resin in the OH− form were obtained. This form was suitable for irradiation since oxygen and hydrogen introduce no interference to iodine analysis by NAA.
About 25 g of Dowex 50X8 (50–100) mesh resins in H+ form were also cleaned by a procedure similar to the one described above, but in this case the final resin was in the NH4 + form. This step was introduced to “clean up” the milk samples from elements such as Na, K, Mg, and Ca which can strongly interfere with the detection of 128I by gamma-ray spectrometry.
Batch separation
Based on the reported affinity of ions for the Dowex (1X8) resin (I− > HSO4 − > ClO3 − > NO3 − > Br− > CN− > HSO3 − > NO2 − > Cl− > HCO3 − > IO3 − > HCOO− > Ac− > OH− > F− [8]), it was hypothesized that iodide and iodate ions could be quantitatively separated if the resin was in the OH− form. To evaluate this hypothesis, a standard solution of 0.5 μg mL−1 of iodine containing 0.25 μg mL−1 of each of iodide and iodate ions was prepared. Approximately 50 mL of the standard solution were stirred with about 0.5 g of Dowex (1X8) resin in OH− form for 5 min and a batch separation was carried out. The sample was filtered and 0.75 mL of the filtrate was used for PC-INAA-CSS. The batch separation was repeated 2 more times. Three spectra were collected and compiled for the quantification of the iodine remaining in solution after each batch separation. The same experiment was then performed using milk samples.
Determination of iodine species in bovine milk
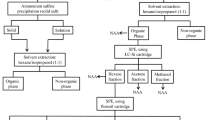
Five replicates of 50-mL of each type of milk samples were analyzed following the separation scheme given in Fig. 1. Once the milk fractions were separated, the irradiation and counting conditions employed for the determination of the iodine bound to each fraction are summarized in Table 1. Sample size in all cases was about 0.75 mL.

Separation scheme for the determination of iodine species in bovine milk
Results and discussion
Neutron activation analysis methods
As mentioned above, four INAA methods were developed in this work for milk analysis. The methods were: INAA using conventional gamma spectrometry and CSS, EINAA using conventional gamma spectrometry, and PC-INAA-CSS. The experimental conditions given in Table 1 were used to analyze milk samples and various separated fractions. A calibration curve (not shown here since it was a straight line) was constructed using 5 replicates of each sample under identical conditions. Slopes of the curves represented the sensitivity for each condition.
The sensitivity, detection limit, precision, accuracy of the four INAA methods were also calculated using two reference materials, namely the NIST Non-Fat Milk Powder SRM-1549 with a certified value of 3.38 ± 0.03 μg g−1 and the NIST Wheat Gluten RM-8418 with a certified value of 0.06 ± 0.01 μg g−1. These materials were analyzed in triplicate and their averages are shown in Table 2. Our measured values agree fairly well with the certified values. A detailed discussion on expanded uncertainties of a PC-EINAA-CSS method has recently been published [17]. It is evident that the highest sensitivity was obtained using INAA and conventional gamma spectrometry under the varied experimental conditions used. The detection limit for each condition was calculated from the analysis of 0.75 mL of homogenized (3.25 % MF) commercial milk sample using Currie’s method [18].
It is also evident from Table 2 that the highest detection limit of 0.1 μg mL−1 was obtained by EINAA-Ge(Li) using a 5–2–10 min irradiation-decay-counting scheme. However, this limit was lowered to 0.02 μg mL−1 using a PC-INAA-CSS method and 6 cycles of irradiation-decay-counting. The improvement in detection limits with increasing number of cycles (N) using the 5–2–10 min scheme in PC-INAA-CSS is shown in Fig. 2. Although the PC-INAA-CSS method provided the lowest detection limit and had to be used when the iodine content was extremely low, the INAA-CSS method was generally sufficient for the determination of total iodine in almost all samples.

Improvement of detection limits with the number of cycles in PC-INAA-CSS
We previously reported that the total iodine content of Canadian bovine milk is about 0.4 μg mL−1 and the organic iodine fraction represents about 10 % of total iodine content [6]. Therefore, three cycles per sample, which represents an attainable detection limit of 0.03 μg mL−1 (Fig. 2), was chosen for detecting iodine species remaining in the filtrate after the batch separation of the inorganic ionic species.
Separation and determination of lipids and proteins
Results of lipid and protein determination in the different milk samples are shown in Table 3. As expected, the lipid content varied directly with the fat content. For both lipids and proteins, the measured values were comparable to those reported on the container by the producer demonstrating the reliability of the methods employed. Lipids were determined quantitatively and the suitability of the solvent system used for the milk lipid extraction has been already reported [6]. Proteins, however, were determined using a very sensitive but relative method (Bradford’s method) whose reliability depended on the standard protein employed. We found the most appropriate standard protein to be used in this case is casein since it represents about 80 % of all milk proteins. The suitability of the ammonium sulfate precipitation for separating milk proteins has also been reported earlier [6].
Inorganic anionic species
Batch separation
The results of the iodine batch separation using the standard iodine solution are shown in Fig. 3. It is evident that two batch separations are sufficient for the extraction of all iodide and iodate ions present in the solution. A plateau at zero counts was attained in Fig. 3 because the iodine standard solution had no other iodine species except the iodide and iodate ions. These experiments show that the OH− form of Dowex (1X8) resin can retain both iodide and iodate anionic species quantitatively.

Batch separation of iodide and iodate ions
Figure 4 shows the results of batch separations in real samples, specifically for homogenized milk (3.25 % MF). All milk samples analyzed exhibited exactly the same behavior. Two batch separations were sufficient for the extraction of all inorganic iodine species from the milk sample. Nevertheless, three extractions were considered as the minimum number required for assuring quantitative separation. In this case, the graph does not reach a plateau at zero. The reason for this observation is that the real milk samples contain organically bound iodine species which are not retained on the resin. In fact, the organic iodine species account for about 10 % of the total iodine present in milk, which agrees with the literature values [6, 8]. It was possible to conclude that a batch separation using Dowex 1X8 (in OH− form) was suitable for the simultaneous separation of both iodide and iodate species from milk.

Inorganic iodine species in homogenized milk by batch separation
The study of the behavior of the inorganic iodide species in milk during the batch separation was carried out by measuring the iodine remaining after each separation. Once the behavior was known and the complete separation of iodide and iodate on the resin was proven, the anionic inorganic iodine in milk was determined by directly irradiating the Dowex (1X8) OH− form resin in the reactor.
Determination of iodine species in bovine milk
The results obtained using the separation scheme (Fig. 1) are presented in Table 4. It appears that the iodine intake due to the consumption of homogenized (3.25 % MF), partly skimmed (2 % MF), partly skimmed (1 % MF) or skim (<0.05 % MF) milk is about the same. However, the consumption of the partly skimmed (1 % MF + Ca) increases the iodine intake by a factor of more than two. Since the inorganic anionic fraction is the one that is significantly increased in this milk, it is possible that the added Ca compound contained iodine and it is retained on the OH− form of the resin. Further studies are however needed to identify the iodine species being added with Ca.
Iodine bound to proteins is essentially the same in all milk samples analyzed in this work. Although there is a smooth variation in the iodine bound to lipids with the MF content as expected, some of these variations perhaps could be attributed to the uncertainty related to the extraction and gravimetric lipid determination techniques.
It is also important to point out that the iodine value reported for each fraction is relative to 1 mL of milk. This value was calculated from the iodine concentration in the separated fraction (e.g. μg g−1 of lipid and protein) multiplied by the lipid and protein content of the milk (g mL−1 milk), respectively. Since the iodine determination in the case of lipid and proteins was carried out in a matrix different than that for total milk and that matrix was mainly composed of light elements (C, H, N, O), a detection limit lower than that reported in Table 2 was achieved.
Conclusions
The iodine levels in lipid, protein and inorganic fractions of commercially available bovine milk with varying fat content were measured with high precision and accuracy by different INAA methods developed here to obtain best detection limits. The total iodine content of milk was determined by INAA-CSS, total organic iodine by PC-INAA-CSS, iodine bound to lipids and proteins by INAA-Ge(Li), and anionic inorganic iodine species by EINAA-Ge(Li).
No significant variation in the iodine content of homogenized (3.25 % MF), partly skimmed (2 % MF), partly skimmed (1 % MF) and skimmed (<0.05 % MF) was observed. Iodine bound to protein was quite constant ranging between 4.8 and 5.0 %. The highest iodine content was found in the anionic inorganic fraction accounting for 90–93 %.
Partly skimmed milk enriched with Ca (1 % MF + Ca), however, had a total iodine concentration of about 2.5 times higher that the rest of the milk types. The inorganic anionic iodine fraction in this milk was also about 2.25 times high. Since some Canadians have been reported to have a daily iodine intake that can be six folds higher than the RNI, perhaps close attention should be given to the consumption of this type of milk although no harmful effect is known so far. One glass (250 mL) of partly skimmed milk enriched with Ca (1 % MF + Ca) per day would mean an iodine intake of about 240 μg which is in excess of RNI of 160 μg and close to the maximum average daily dietary intake (ADDI) of 300 μg iodine recommended by WHO during pregnancy and lactation. Obviously, a glass of milk is not the only food an adult consumes in a day. Moreover, salts in Canada are iodized. Therefore, a cautionary note on iodine intake is well deserved.
References
Breaking barriers not bones (2008) National report card on osteoporosis care. International Osteoporosis Fundation. http://www.iofbonehealth.org. Accessed 21 Sept 2010
Canadian dairy association center. http://www.dairyinfo.gc.ca/index_e.php. Accessed 21 Sept 2010
International dairy federation bulletin (423/2007). http://www.fil-idf.org/Content/Default.asp. Accessed 21 Sept 2010
Borucki Castro SI, Berthiaume R, Laffey P, Fouquet A, Beraldin F, Robichaud A, Lacasse P (2010) Iodine concentration in milk sampled from canadian farms. J Food Prot 73:1658–1663
Zimmermann MB (2008) Iodine requirements and the risks and benefits of correcting iodine deficiency in populations. J Trace Elem Med Biol 22:81–92
Isaac-Olive K, Acharya R, Chatt A (2008) Fractionation analysis of iodine in bovine milk by preconcentration neutron activation analysis. Talanta 77:827–832
Leiterer M, Trukenbrodt D, Franke K (2001) Determination of iodine species in milk using ion chromatographic separation and ICP-MS detection. Eur Food Res Technol 213:150–153
Hou X, Dahlgaard H, Jacobsen U, Nielsen SP (2000) Pre-separation neutron activation analysis of seawater, urine and milk for iodide and iodate. J Radioanal Nucl Chem 244:87–91
Bhagat PR, Pandey AK, Acharya R, Nair AGC, Rajurkar NS, Reddy AVR (2007) Selective preconcentration and determination of iodine species in milk samples using polymer inclusion sorbent. Talanta 71:1226–1232
Sanchez LF, Szpunar J (1999) Speciation analysis for iodine in milk by size-exclusion chromatography with inductively coupled plasma mass spectrometric detection (SEC-ICP MS). J Anal At Spectrom 14:1697–1702
Fernández-Sánchez LM, Bermejo-Barrera P, Fraga-Bermudez JM, Szpunar J, Lobinski R (2007) Determination of iodine in human milk and infant formulas. J Trace Elem Med Biol 21(Suppl 1):10–13
Michalke B (2006) Trace element speciation in human milk. Pure Appl Chem 78:79–90
Rivero Martino FA, Fernández Sánchez LM, Sanz Medel A (2002) Multi-elemental fractionation in milk whey by size exclusion chromatography coupled on line to ICP-MS. J Anal At Spectrom 17:1271–1277
Ryan DE, Stuart DC, Chattopadhyay A (1978) Rapid multielement neutron activation analysis with a slowpoke reactor. Anal Chim Acta 100(C):87–93
Stuart DC, Ryan DE (1981) Epithermal neutron activation analysis with a SLOWPOKE nuclear reactor. Can J Chem 59:1470–1475
Holzbecher J, Chatt A, Ryan DE (1985) SLOWPOKE epi-cadmium neutron flux in activation analysis for trace elements. Can J Spectrosc 30:67–72
Acharya R, Chatt A (2009) Determination of iodine in biological materials by pseudo-cyclic epithermal INAA using anti-coincidence gamma-ray spectrometry and estimation of expanded uncertainties. J Radioanal Nucl Chem 282:991–996
Currie LA (1968) Limits for qualitative detection and quantitative determination—application to radiochemistry. Anal Chem 40:586–593
Acknowledgments
The authors would like to acknowledge the financial assistance from the (i) Natural Sciences and Engineering Research Council (NSERC) of Canada in the form of Research Operating/Discovery and Major Facilities Access grants, (ii) Dalhousie University for the DUSR Facility Operating Grant, and (iii) Faculty of Graduate Studies for a scholarship (KI-O). The authors are grateful to Dr. J. Holzbecher of the DUSR facility for assistance in irradiations, and to the High Institute of Nuclear Science and Technologies, Ministry of Science of the Government of Cuba for a study leave (KI-O).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Isaac-Olive, K., Chatt, A. Studies of total, organic and inorganic iodine in Canadian bovine milk samples with varying milk fat content using ion-exchange chromatography and neutron activation analysis. J Radioanal Nucl Chem 294, 479–486 (2012). https://doi.org/10.1007/s10967-012-1849-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-012-1849-0