
Abstract
Radiolabeling of oxybutynin, a muscarinic acetylcholine (mACh) receptor antagonist agent with 99mTc is of considerable interest for imaging of urinary bladder. This study is aimed to optimize radiolabeling yield of oxybutynin with 99mTc using SnCl2·2H2O as a reducing agent with respect to factors that affect the reaction conditions such as oxybutynin amount, stannous chloride amount, reaction time and pH of the reaction mixture. In vitro stability of the radiolabeled complex was checked and it was found to be stable for up to 8 h. 99mTc-oxybutynin was injected via subcutaneous and intravenous administration routes into normal Sprague–Dawley rats. Biodistribution studies have revealed that 99mTc-oxybutynin exhibits high affinity and specificity for the muscarinic M3 subtype located on the smooth muscle of urinary bladder relative to the M1 and M2 subtypes of the G protein coupled receptor (GPCR) superfamily. In vivo uptake of subcutaneous 99mTc-oxybutynin in urinary bladder was 19.6 ± 0.42% ID at 0.5 h, whereas in intravenous administration route the accumulation in the urinary bladder was found to be 9.4 ± 0.31% ID at 0.5 h post injection. Administration of cold oxybutynin effectively blocked urinary bladder uptake and further confirms the high specificity of this complex for the M3 receptor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
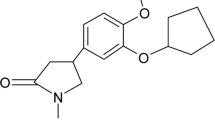
Oxybutynin, 4-Diethylaminobut-2-ynyl-2-cyclohexyl-2-hydroxy-2-phenyl-ethanoate (Fig. 1), is a muscarinic acetylcholine (mACh) receptor antagonist agent developed as an antispasmodic, anticholinergic drug for the treatment of symptoms of overactive bladder (OAB) such as urge urinary incontinence [1]. Consequently, relaxes bladder smooth muscle in patients with conditions characterized by involuntary bladder contractions [2, 3]. The urinary bladder is under the control of the parasympathetic nervous system, which classically releases acetylcholine (ACh). ACh in turn acts via muscarinic receptors located on the detrusor smooth muscle of the urinary bladder to evoke contraction of the bladder smooth muscle and so voiding of urine [4].

Chemical structure of oxybutynin
The muscarinic acetylcholine (mACh) receptor family consists of five members and belongs to the G protein-coupled receptor (GPCR) superfamily [5, 6]. The five mACh receptor subtypes have been identified and classified (M1–M5) based on genetic and pharmacological characteristics [7–9] and genetic/gene knockout [10–12]. Although both M2 and M3 subtypes coexist in smooth muscle, muscarinic M3 receptors are primarily responsible for the contraction of most smooth muscle, including the urinary bladder [13]. The M2 receptors may have a role in mediating indirect contractions and/or inhibition of detrusor relaxation [14]. In vitro radioligand studies with human recombinant muscarinic subtypes have revealed that oxybutynin exhibits high affinity and specificity for the muscarinic M3 subtype located on the smooth muscle of urinary bladder relative to the M1 and M2 subtypes [15, 16]. On the basis of these results, mACh receptor antagonists with a higher affinity for M3 than M2 subtypes should be more beneficial in the treatment of overactive bladder. Different reviews have explored the binding of ligand to receptors with particular focus on the urinary bladder in the presence of various pharmacokinetic and pharmacodynamic factors in order to treat OAB [17–21].
Muscarinic receptor binding has been characterized and measured in the urinary bladder. Therefore, numerous studies involving antimuscarinic agents to treat OAB have revealed their distribution, function with respect to extent and duration of receptor binding at target and non-target tissues after clinical administration of oxybutynin [22–26]. The characterization of muscarinic receptor binding is an extremely powerful way to elucidate in vivo pharmacological specificity, such as organ selectivity, efficacy, and safety, in relation to the pharmacokinetics and pharmacodynamics [27]. The effectiveness of antimuscarinic agents in the treatment of the overactive bladder (OAB) is also thought to arise through blockade of bladder muscarinic receptors located on detrusor smooth muscle cells [28].
Oxybutynin is metabolized primarily by the cytochrome P450 enzyme systems, particularly CYP3A4 found mostly in the liver and gut wall [29, 30]. Its metabolic products include phenylcyclohexylglycolic acid, which is pharmacologically inactive, and N-desethyloxybutynin (DEOB), which is pharmacologically active but does not substantially influence the clinical efficacy of oxybutynin. DEOB contributes to the high incidence of anticholinergic adverse effects [31, 32]. Subcutaneous administration of oxybutynin efficiently avoids first-pass hepatic metabolism and conversion of oxybutynin to desethyloxybutynin [33, 34].
Bladder cancer is the fourth most common type of cancer in the United States and the fifth in Europe [35]. In developing countries, 75% of cases are squamous cell carcinomas caused by Schistosoma haematobium (bilharzia) infection [36, 37]. Bladder cancer accounted for approximately 90% of cancers of the urinary collecting system (renal pelvis, ureters, bladder, and urethra) [38]. It usually originates in the bladder lining, which consists of a mucous layer of surface cells that expand and deflate (transitional epithelial cells), smooth muscle, and a fibrous layer [39]. About 80% of bladder tumors are classified as superficial (bladder mucosa), and 20% as muscle invasive. The treatment of bladder cancer depends on how deep the tumor invades into the bladder wall. Superficial tumors are fairly benign, and invasive tumors are highly malignant [40].
Hyaluronic acid (HA)-paclitaxel bioconjugate was labeled with 99mTc [41] to target superficial bladder cancer. Its biodistribution was evaluated after intravenous (uptake = 4.87 ± 0.64), intraperitoneal (1.21 ± 1.05), intravesical (95.03 ± 5.51) and oral (0.1% ± 0.05) administration routes in mice. The bioavailability studies conducted for 99mTc-Ha-paclitaxel showed that the intravesical and oral administration routes of the complex could be used for local treatment of bladder or stomach superficial cancers, while the intraperitoneal route might be useful against tumours spreading within the peritoneal cavity [41]. The complex administered intravenously could be used for liver metastasis therapy due to its high physiological and receptor-specific liver uptake.
The aim of the present work was to establish a simple and efficient method for radiolabeling oxybutynin with 99mTc as a urinary bladder imaging agent.
Experimental
Chemicals
Oxybutynin chloride (USP) was purchased from Sigma–Aldrich (USA). All other chemicals and solvents were of analytical reagent grade and were used directly without further purification unless specified. Distilled water was used in all experiments for the preparation of solutions, dilution and washing purposes.
Radiolabeling procedure
A specific amount of oxybutynin (5 mg) dissolved in 0.5 ml deoxygenated water, 100 μg of freshly prepared deoxygenated aqueous solution of stannous chloride dihydrate and 0.5 ml sodium pertechnetate (400 MBq) were introduced. The pH of the reaction mixture was adjusted to 4 using phosphate buffer and the preparation was mixed in sterile glass vial and under positive nitrogen gas pressure, closed with a rubber stopper and an aluminum cap. The mixture was agitated in a vortex mixer and left to react at room temperature (25 °C) for 30 min. The factors that affect the labeling yield such as oxybutynin amount (1–10 mg), stannous chloride amount (10–150 μg), reaction time (5–120 min) and pH of the reaction mixture (2–9) were examined to optimize the reaction conditions. 99mTc-oxybutynin complex was filtered through 0.22 μm Millipore filter to separate the colloid and it was further purified by reversed phase-HPLC (RP-HPLC) for in vivo biodistribution studies.
Analysis of 99mTc-oxybutynin complex
Radiochemical purity of 99mTc-oxybutynin was performed by thin layer chromatographic method using strips of silica gel impregnated glass fiber sheets (ITLC-SG). Free 99mTcO4 − in the preparation was determined using acetone as the mobile phase. Reduced hydrolyzed technetium was determined by using 5 N HCl as the mobile phase. It was further confirmed by a Shimadzu HPLC system, which consists of pumps LC-9A, UV spectrophotometeric detector operated at a 220 nm (SPD-6A), and rheodyne injection valve. Chromatographic analysis of 99mTc-oxybutynin was performed by injection of 10 μl of the reaction mixture at the optimum conditions into a reversed-phase column, (Waters, Symmetry C18; 5 μm, 4.6 mm × 150 mm) preceded by a guard column (Waters, Symmetry C18; 5 μm) and eluted with mobile phase consisting of acetonitrile and 0.01 M potassium dihydrogen phosphate/diethylamine (60:40:0.2 v/v) [42]. The mobile phase was filtered and degassed prior to use and the flow rate was 0.5 ml/min.
Stability of 99mTc-oxybutynin in serum
Stability of 99mTc-oxybutynin complex in serum was studied in vitro by mixing 1.8 ml of normal serum and 0.2 ml of 99mTc-complex and incubated at 37 °C for 24 h. Exactly 0.2 ml aliquots were withdrawn during the incubation at different time intervals up to 24 h and subjected to ITLC for determination the percent of 99mTc-complex, reduced hydrolyzed technetium and free pertechnetate. Consequently, the stability of the radiolabeled complex will determine its suitability for in vivo application.
Biodistribution studies of 99mTc-oxybutynin in normal rats
The experimental procedures of the animal studies were in accordance with the guidelines set out by the Egyptian Atomic Energy Authority and were approved by the animal ethics committee, Labeled Compound Department. Biodistribution studies of 99mTc-oxybutynin in normal Sprague–Dawley rats (n = 5), were carried out at 30 min, 1 and 4 h for both subcutaneous and intravenous administration routes. Prior to the study, animals were housed in groups of five and provided with food and water. The animals were anesthetized with diethyl ether and shaved in the upper back. Aliquots of 100 μl containing 18 MBq of the purified labeled complex were injected into each animal via the tail vein or subcutaneously to avoid the first-pass effects of liver and gut wall. Each animal was housed individually before sacrifice by cervical dislocation at 30 min, 1 and 4 h after administration of 99mTc-oxybutynin (n = 5). Each rat was weighed and the blood drawn from the heart and weighed immediately following sacrifice. After dissection, the organs and tissues were rinsed with saline, collected in plastic containers and weighed. The radioactivity of each sample as well as the background was counted in a well-type NaI(Tl) well crystal coupled to SR-7 scaler ratemeter. Urinary bladders were removed from Sprague–Dawley rats weighing 150–170 g. Bladder was emptied and the urine from the bladder, kill paper and from the cage paper was collected and counted. The bladder muscle was counted and the percent-injected dose per organ (% ID/organ ± SD) in a population of five rats for each time point for both subcutaneous and intravenous administration routes are reported. Differences in the data were evaluated with the Student t test. Results for P using the 2-tailed test are reported and all results are given as mean ± SEM. The level of significance was set at P < 0.05.
Results and discussion
Radiochemical purity and stability of 99mTc-oxybutynin complex was assessed by thin layer chromatographic method and reversed-phase high-performance liquid chromatography (HPLC). In thin layer chromatography using acetone as the solvent, free99mTcO4 − moved with the solvent front (R f = 1), while 99mTc-oxybutynin and reduced hydrolyzed technetium remained at the origin. Reduced hydrolyzed technetium was determined by using 5 N HCl as the mobile phase, where reduced hydrolyzed technetium remains at the origin (R f = 0) while other species migrate with the solvent front (R f = 1). The radiochemical purity was determined by subtracting the sum of the percent of colloid and free pertechnetate from 100%. On the other hand, the HPLC purification method separated radiolabeled complex from the unlabeled compound and the free pertechnetate.99mTc-oxybutynin complex was collected at retention time 3.9 min, the free pertechnetate was separated at 2.6 min and the oxybutynin at ~3 min as shown in Fig. 2. The experimental results after purification showed that the radiochemical purity was more than 98%.

RP-HPLC chromatogram of 99mTc-oxybutynin
Effect of ligand concentration
As shown in Fig. 3, the labeling yield of 99mTc-oxybutynin complex was 85.3% at 1 mg oxybutynin and increased with increasing the amount of oxybutynin till reaching the maximum value of 93.5% at 5 mg. The amount of oxybutynin at 5 mg was sufficient enough toward the formation of 99mTc-oxybutynin complex with high labeling efficiency. The labeling efficiency remained stable even with increasing the amount of oxybutynin above 5 mg.

Effect of oxybutynin amount on the labeling yield of 99mTc-oxybutynin
Effect of SnCl2·2H2O concentration
The effect of the amount of stannous chloride was summarized in Fig. 4. The data show that the radiochemical yield was dependent on the amount of SnCl2·2H2O present in the reaction mixture. At 10 μg SnCl2·2H2O, the labeling yield of 99mTc-oxybutynin was 82.1% due to SnCl2·2H2O concentration was insufficient to reduce all pertechnetate so the percentage of 99mTcO4 − was relatively high (14.9%). The labeling yield was significantly increased by increasing the amount of SnCl2·2H2O in the range of 10–100 μg, at which maximum labeling yield of 93.5% was obtained. By increasing the amount of SnCl2·2H2O above the optimum concentration value, the labeling yield decreased because excess SnCl2·2H2O was converted to colloid (13.2% at 150 μg SnCl2).

Effect of SnCl2·2H2O concentration on the labelling yield of 99mTc-oxybutynin; reaction conditions: 5 mg oxybutynin, x μg of SnCl2·2H2O, 0.5 mL (~400 MBq) of 99mTcO4 − at pH 4, the reaction mixture was kept at room temperature for 30 min
Effect of pH
Radiochemical yield of 99mTc-oxybutynin complex was affected by changes in pH as graphically illustrated in Fig. 5. The maximum yield was obtained at pH 4. At pH 2, the radiochemical yield was relatively low (89.1%) with the appearance of colloids as predominant species compared to 65.2% at pH 9 with the formation of colloids. At lower pH (2–4), the reducing power of Sn(II) ion works strongly in acidic medium to reduce all pertechnetate to TcO(V), which favor the formation of 99mTc-oxybutynin. The labeling yield decreased to 65.2% at higher pH due to the destruction of the reductive ability of SnCl2, which decreased the labeling yield.

Effect of pH on the labeling yield of oxybutynin with 99mTc; reaction conditions: 5 mg oxybutynin, 100 μg of SnCl2·2H2O, 0.5 mL (~400 MBq) of 99mTcO4 − at pH = x, the reaction mixture was kept at room temperature for 30 min
Effect of reaction time
Figure 6 describes the labeling yield of 99mTc-oxybutynin complex at different reaction times. The labeling yield increased with time till reaching 93.5% at 30 min. It remained constant (~94%) even when the reaction time was extended to 8 h.

99mTc-oxybutynin yields vs. reaction time; reaction conditions: 5 mg oxybutynin, 100 μg of SnCl2·2H2O, 0.5 mL (~400 MBq) of 99mTcO4− at pH 4, the reaction mixture was kept at room temperature for different intervals of time
Stability in human serum
Human serum stability is a method used for the evaluation of the stability of radiolabeled complexes in vitro to probe their stabilities and behavior in vivo over time. Incubation of the preparation containing 99mTc-oxybutynin in normal serum up to 24 h at 37 °C resulted in a slight decrease of the labeling yield to 92.2% i.e., small release of radioactivity (1.3%, n = 5 experiments) from the 99mTc-oxybutynin complex as determined by ITLC.
Biodistribution
Biodistribution studies are shown in Table 1 as average percent-injected dose per organ (% ID/organ) in a population of five rats per time point. The biodistribution results demonstrated that the urinary bladder uptake of the 99mTc-oxybutynin was found to be 19.6 ± 0.4% and 9.4 ± 0.2% for subcutaneous and intravenous, respectively, at 0.5 h post injection. The intravenous uptake of 99mTc-oxybutynin was greater than that of 99mTc-Ha-paclitaxel bioconjugate (uptake = 4.34%) in the bladder [41], which reflected receptor-specific bladder uptake for 99mTc-oxybutynin.
Retention of the radiopharmaceutical in urinary bladder decreased to 3.2 ± 0.2% and 2.3 ± 0.1% for subcutaneous and intravenous injection, respectively, at 4 h post injection. 99mTc-oxybutynin was cleared efficiently from the blood stream with only 0.7–1% remaining in the blood after 4 h post injection in comparison to 7–9% ID at 0.5 h post injection and is washed out through the kidneys and liver within 4 h post injection. The hepatobiliary excretion through the intestines (44% ID) is due to the lipophilic nature (Log P = 4.2) [43, 44] of the oxybutynin that would increase hepatic excretion (liver and intestines) despite a significant amount of the radioactivity was excreted via the renal-urinary system (36% ID).
Blocking studies, in which cold oxybutynin was administered 30 min prior to the administration of 99mTc-oybutynin complex, reduced the percent uptake in urinary bladder at 30 min post injection to 3.9 ± 0.5 and 1.3 ± 0.0% for subcutaneous and intravenous administration routes, respectively (Fig. 7). Administration of cold oxybutynin effectively blocked urinary bladder uptake reflecting the high specificity this complex has for the GPCR receptors.

In vivo blocking vs. non blocking in urinary bladder for 99mTc-oxybutynin at 0.5 h post injection
Conclusions
Oxybutynin was labeled with 99mTc by direct labeling method at room temperature with a high labeling yield of 93.5% using stannous chloride as a reducing agent. This study described the in vitro and in vivo characterization of 99mTc-oybutynin complex necessary for designing a potentially useful radiopharmaceutical for the M3-mediated diagnostic imaging of the urinary bladder. It showed good radiochemical and metabolic stability in vivo. Receptor mediated urinary bladder uptake of 19.56% confirmed the effectiveness of this complex for targeting GPCR receptor-specific tissue.
References
Andersson KE (2004) Lancet Neurol 3:46–53
Bulmer P, Abrams P (2000) Pharmacother 11:1–11
Resnick NM, Yalla SV (1985) New Eng J Med 313:800–805
Jonathan JS, Dmochowski RR (2006) Rev Urol 8(3)
Hulme EC, Birdsall NJM, Buckley NJ (1990) Annu Rev Pharmacol Toxicol 30:633–673
Caulfield MP (1993) Pharmacol Ther 58:319–379
Hegde SS, Choppin A, Bonhaus D, Briaud S, Loeb M, Moy TM, Loury D, Eglen RM (1997) Br J Pharmacol 120:1409–1418
Sawyer GW, Ehlert FJ (1999) J Pharmacol Exp Ther 289:464–476
Ehlert FJ, Sawyer GW, Esqueda EE (1999) Life Sci 64:387–394
Stengel PW, Gomeza J, Wess J, Cohen ML (2000) J Pharmacol Exp Ther 292:877–885
Stengel PW, Yamada M, Wess J, Cohen ML (2002) Am J Physiol 282:R1443–R1449
Matsui M, Motomura D, Karasawa H, Fujikawa T, Jiang J, Komiya Y, Takahashi S, Taketom M (2000) Proc Natl Acad Sci USA 97:9579–9584
Eglen RM, Reddy H, Watson N, Challis RAJ (1994) Trends Pharmacol Sci 15:114–119
Maruyama S, Oki T, Otsuka A, Shinbo H, Ozono S, Kageyama S, Mikami Y, Araki I, Takeda M (2006) J Urol 175:365–369
Ikeda K, Kobayashi S, Suzuki M, Miyata K, Takeuchi M, Yamada T, Honda K (2002) Naunyn Schmiedebergs Arch Pharmacol 366:97–103
Kobayashi S, Ikeda S, Miyata K (2004) Life Sci 4:843–853
Beauchamp HT, Chang RSL, Sigel PKS, Gibson RE (1995) J Pharmacol Exp Ther 272:612–618
Uchida S, Yamada S, Ohkura T, Heshikiri M, Yoshimi A, Shirahase H, Kimura R (1995) Br J Pharmacol 114:217–223
Ohkura T, Yamada S, Deguchi Y, Kimura R, Matsushima H, Higuchi S, Inagaki O, Honda K, Takenaka T (1998) Life Sci 63:2147–2155
Oki T, Kimura R, Saito M, Miyagawa I, Yamada S (2004) J Urol 172:2059–2064
Nilvebrant L, Andersson KE, Gillberg PG, Stahl M, Sparf B (1997) Eur J Pharmacol 327:195–207
Abrams P, Freeman R, Anderstrom C, Mattiasson A (1998) Br J Pharmacol 81:801–810
Anderson RU, Mobley D, Blank B, Saltzstein D, Susset J, Brown JS (1999) J Urol 161:1809–1812
Chapple CR (2000) J Urol 55:33–46
Davila GW, Daugherty CA, Sanders SW (2001) J Urol 166:140–145
Gupta SK, Sathyan G (1999) J Clin Pharmacol 39:289–296
Oki T, Okura AT, Yamada S (2005) J Pharmacol Exp Ther 316(3)
Abrams P, Andersson KE, Buccafusco JJ, Chapple C, de Groat CW, Fryer AD, Gary K, Laties A, Nathanson NM, Pasricha PJ, Wein AJ (2006) Br J Pharmacol 148:565–578
Douchamps J, Derenne F, Stoickis A, Gangji G, Juvent M, Herchuelz A (1998) Eur J Clin Pharmacol 35:515–520
Lukkari E, Taavitsainen P, Juhakoski A, Pelkonen O (1998) Pharmacol Toxicol 82(4):161–166
Dmochowski RR, Sand PK, Zinner NR, Gittelman MC, Davila GW, Sanders SW (2003) J Urol 62:237–242
Uchida M, Yamada S (2005) Life Sci 76:2445–2456
Waldeck K, Larsson B, Andersson KE (1997) J Urol 157:1093–1097
Yaïch M, Popon M, Médard Y, Aigrain E (1998) Pharmacogenetics 8(5):449–451
Metts MC, Metts JC, Milito SJ, Thomas CR (2000) J Natl Med Assoc 92(6):285–294
Khurana S, Dubey ML, Malla N (2005) Indian J Med Microbiol 23(2):74–79
Ross AGP, Bartley PB, Sleigh AC, Olds GR, Li Y, Williams GM, McManus DP (2002) N Engl J Med 346:1212
Millar J, van der Meijden APM (1999) Br Med J 318:875a–875
Horwich A (2000) N Engl J Med 342:1297
van der Meijden APM (1998) Br Med J 317:1366–1369
Alafort LM, Riondato M, Nadali A, Banzato A, Camporese D, Boccaccio P, Uzunov N, Rosato A, Mazzi U (2006) J Label Compd Radiopharm 49:939–950
El-Gindy A (2005) IL Farmaco 60:689–699
Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L (2007) J Chem Inf Model 47:2140–2148
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moustapha, M.E., Motaleb, M.A. & Ibrahim, I.T. Synthesis of 99mTc-oxybutynin for M3-receptor-mediated imaging of urinary bladder. J Radioanal Nucl Chem 287, 35–40 (2011). https://doi.org/10.1007/s10967-010-0794-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-010-0794-z