
Abstract
Sorption of Cs, Pu and Am on natural clay of complex composition was studied to better understand the sorption mechanisms. It was found that cesium sorption to natural clay was affected by its coatings and by the ionic strength of solution. The sorption of Pu and Am on the clay was compared with that on synthetic goethite, hematite and magnetite, representing components of the clay coatings. The sorption was quantitatively interpreted using models assuming ion exchange and/or complex formation on the “layer sites” and “edge sites” of the clay and its coatings. Constants characterizing properties of the sites and sorption equilibria were determined.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Natural clay minerals are locally available low cost materials which are often used for engineered barriers. The complex composition of natural minerals can be considered as their advantage since radionuclides with a wide range of chemical properties can find appropriate sorption sites on their surfaces. On the other hand, the sorption processes are rather complicated in these heterogeneous systems and they can be affected by many factors and for this reason they are difficult to predict. The knowledge how the mineralogical composition of clay minerals and their coatings can influence retention of radionuclides is an important issue for the performance assessment. 137Cs, 241Am and Pu isotopes belong to radionuclides sorption and migration of which from radwaste repositories are most often studied. It is generally recognized that Cs+ is strongly and selectively sorbed by the phyllosilicate fraction of soils, sediments and suspended particles and its selective adsorption by mica-like minerals, such as illite, can considerably reduce its movement and adverse effects in the environment [1, 2]. The major sorbents for cesium are the layer-type silicates that bind Cs either through electrostatic attraction of hydrated Cs+ by negative charges within the basalt plane and dissociated edge hydroxyl groups forming outer-sphere complexes or through electronic bonding at the frayed edge sites (FES), external basalt sites or within the interlayer forming inner-sphere complexes [3]. The high selectivity FES sites were determined in many studies and it was found that they constitute only a small part of about 0.02% of the total cation exchange capacity [4]. Cesium adsorption by layer-type silicates has been often described as a cation exchange process on two or more sites with distinctly different selectivity.
The most important components of natural mineral surface coatings affecting adsorption of radionuclides are manganese oxides, iron oxides and organic substances [5]. The retention of Cs by iron oxides such as goethite, hematite and magnetite was found to be negligible [6]. Naturally occurring organic substances (humic substances, polysaccharides and proteins) can affect various adsorption sites in different ways: by preventing clay layer collapse at frayed edge sites, resulting in enhanced Cs sorption; or by inhibiting Cs sorption to clay minerals via modification of the properties of regular exchange sites [7]. Complexation ability of natural soil organics towards Cs was found to be rather weak [8].
On the other hand, humic substances (HS) are widely recognized as important complexing ligands for actinide in aquatic environments [9]. However, quantitative description and prediction of the complexation with HS are rather difficult due to the complex nature of HS composed of a variable mixture of molecules with a number of carboxylic and phenolic functional groups. Interpretation of the interaction of tetravalent actinides with HS was improved when consistent sets of hydrolysis constants for the actinides were taken into account [10]. The important role of natural organic substances in the transformation of Pu oxidation state was corroborated in recent publications [11, 12].
Rather contradictory data on the sorption behavior of plutonium and americium in the environment have been published [13–15]. In soil and bottom sediment samples, Pu isotopes were found to be associated with carbonates, Fe and Mn oxides as well as organic substances [16, 17]. Short time sorption experiments and geochemical modeling indicated that sorption of Pu was controlled by a competition between complexation with iron oxyhydroxides and the precipitation of hydrolysis products [18].
In order to contribute to the knowledge of the mechanisms of radiocesium, plutonium and americium sorption on natural clays and their coatings, we studied the sorption under laboratory conditions. The experimental data were modeled using ion exchange and surface complexation models.
Materials and methods
Triassic clay selected as engineered barrier for the cement (concrete) based near surface low and intermediate level radioactive waste repository was obtained from the industrial exploitation site Šaltiškiai in North Lithuania. Chemical composition of the clay is SiO2 45.51%, Al2O3 13.50%, Fe2O3 5.17% MgO 3.00%, CaO 12.88%, Na2O 0.28%, K2O 5.02%, TiO2 0.43%, total S 0.16%, loss on ignition 13.96%. Triassic clay is composed of micro-aggregates of clay particles (with 56–71% of smectite, including 14% of montmorillonite, 20% of illite and from 1 to 9% of chlorite minerals) which are cemented by limonite FeO(OH)·nH2O. Two samples of the clay were used for the sorption studies: sample S(1) contained siderite (FeCO3 1.6%), hematite (α-Fe2O3 2.3%), goethite (α-FeOOH 0.1%), calcite (CaCO3 19.8%) and total organic carbon (TOC 0.034%) in surface coatings and sample S(2)—originally S(1) from which surface coatings containing carbonates and iron oxides were removed by leaching with 2 mol/L HCl and then by citrate-dithionite treatment [19].
In addition, sorption experiments were conducted with synthetic iron oxides for better understanding of their role in the sorption processes on the clay. The iron oxides (goethite, hematite and magnetite) were synthesized using procedures described in literature [20–22] and characterized by means of Mössbauer spectroscopy [23]. Details of the physical and mineralogic characteristics of all the solids used in these studies were reported previously [24]. Surface properties of the solids [point of zero charge (pHpzc), acid–base characteristics of their surface sites, etc.] were determined by acid–base titrations using a slight modification of the technique described by Wanner et al. [25].
In all experiments ACS reagent grade or higher grade chemicals were used. All solutions were freshly prepared using deionized (Elix, Milipore) or Milli-Q (Millipore Milli-Q Synthesis A-10) water.
The standard laboratory batch method was used in all sorption experiments [26]. Sorption was studied (at 25 °C under Ar atmosphere) from 0.01 or 0.1 mol/L NaNO3 solutions, which were mixed with the solids at solid to solution ratio of 1 g/L in polypropylene bottles. pH of suspensions thus obtained was adjusted with nitric acid or sodium hydroxide and repeated wash with fresh portions of the working solution was performed until required pH remained stable of over ±0.1 pH units for 48 h and then 134Cs, Pu (mixture of isotopes isolated from the Chernobyl soil) [23, 24] or 241Am spikes were added to achieve their desired initial concentration. pH was measured using WTW pH-electrode SenTix 41or SenTix 81 before and after sorption experiments.
The initial concentration of cesium in solutions was 6.80 × 10−5 mol/L. The oxidation state purity of Pu(IV) stock solution was analyzed by solvent extraction at pH 0.5 using 0.5 M thenoyl-trifluoro-acetone (TTA) as extractant. Typically 96 ± 3 % of the total plutonium was found in the tetravalent state. Starting concentrations of Pu(IV) and 241Am were 1 × 10−10 and 3 × 10−11 mol/L, respectively.
Suspensions were shaken for time sufficient for establishment of sorption equilibrium and then the solids were separated from solution by centrifugation at 10,000–20,000g. Plutonium in the solution and solid phase was determined after radiochemical separation using the UTEVA and TRU (Eichrom Industries). Pu and Am activities were measured by alpha spectrometry. 242Pu and 243Am were used as tracers in the separation procedure. 134Cs activities were measured with an intrinsic germanium detector. Adsorption losses of the radionuclides from solutions to polypropylene bottle walls varied from 0.1 to 1% of the total radionuclide present and decreased with an increase of the sorption time. They were taken into account in calculations.
Results and discussion
Cs(I), Pu(IV) and Am(III) sorption on clay samples and synthetic iron oxides representing components of the clay coatings was studied from NaNO3 solutions as a function of pH. The data obtained, presented in Figs. 1, 2, 3, 4 and 5, were interpreted using suitable sorption models assuming three kinds of sorption mechanisms, namely ion exchange on layer sites XH and ion exchange or surface complex formation on deprotonated edge sites SO−. Terms “layer sites” and “edge sites”, common for clays, were also used for clay coatings, composed of oxides with different structure of crystals. This distinction facilitates evaluation of acid–base titrations. Assumption of existence of the layer sites on magnetite already enabled modeling of Cs and Sr sorption on this mineral [27]. Best fitting of the experimental data by the model was sought, and parameters (equilibrium constants) characterizing individual sorption mechanisms were determined. The models consisted of sets of equations describing protonation/deprotonation of the surface sites, ion exchange and simple sorption reactions shown in Table 1, together with corresponding mass balance equations. Choice of the sorption reactions was based on the calculated speciation of studied radionuclides in solution. The corresponding computer codes were constructed in software product Famulus using the Newton–Raphson multidimensional nonlinear regression method.

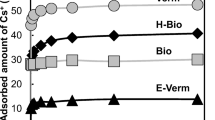
pH dependences of Cs sorption to clay samples from 0.01 and 0.1 mol/L NaNO3, experimental and modeling data

Mole fraction (F) of active sites on the clay surface (XH, layer sites; SOH0, SOH2 + and SO−, edge sites; ionic strength = 0.1)

Diagram of Pu(IV) and Am(III) aqueous speciation

Data on the sorption of Pu(IV) and Am(III) (%) on clay S(1) fitted with sorption models and compared with calculated speciation of Am and Pu on the solids. [Clay] = 64.3 m2/L

Data on the sorption of Pu(IV) and Am(III) (%) on iron oxides: experimental and modeling results. [Goethite] = 172 m2/L, [Hematite] = 192 m2/L, [Magnetite] = 114 m2/L
The first step in the modeling was determination of the mass concentration (in mol/kg) of the layer sites ΣXH, of the edge sites ΣSOH (all forms) and their characteristic constants (KX1, KS1 and KS2) for corresponding solid by acid–base titration. Changes in the concentration of the particular sorption sites on the surface of clay particles can be calculated from the obtained data (Table 1, 2). Graphical presentation of clay S(1) active sites available for sorption process is given in Fig. 2. The calculated parameters were used as input data to the sorption codes. The fitting of sorption data proceeded in the iteration cycle, from which it was possible to determine the sought parameters when the difference of the sum of relative squares of deviations after two successive cycles (i.e. ith and ith + 1) was <10−8. As a fitting criterion, reflecting the agreement between calculated and experimental values, the WSOS/DF (weighted sum of squares divided by the degrees of freedom) quantity was calculated [28]. Generally, the agreement is acceptable if WSOS/DF ≤ 20.
In the surface complexation modeling (SCM) of Cs, Pu and Am sorption on edge sites, three types of SCMs, namely two electrostatic models, i.e. constant capacitance (CCM) and diffuse double layer (DLM) model, and one chemical equilibrium, non-electrostatic model (CEM), were employed. The best fit was found with CEM. Ion exchange of Cs on layer sites was modeled by the IExM model. The resulting values of equilibrium constants are summarized in Tables 1 and 2. According to the obtained values of the fitting criterion (WSOS/DF varied from 0.05 to 20.3), agreement between the calculated and experimental sorption values was very good. Only in the case of Am sorption on goethite resulted in WSOS/DF 29.4. The goodness-of-fit is also demonstrated in Figs. 1, 2, 3, 4 and 5, where curves calculated using the constants from Table 1 represent the best fit of the corresponding sets of experimental data.
In the case of cesium sorption on clay samples S(1) and S(2), the results shown in Fig. 1 indicate large effect of the removal of surface coatings from the clay and of the ionic strength of the solution. The sorption from 0.01 mol/L NaNO3 on the untreated clay S(1) can be quite well modeled assuming two sorption mechanisms. At pH < 6, ion exchange on the layer sites predominates, whereas also ion exchange on the edge sites takes place at pH 7–10. The edge sites are probably located on clay coatings. That is why ion exchange on the edge sites practically disappears when the coatings are removed [clay S(2)]. The large deviation of the experimental data from the modeling results at pH 4–6 can be explained either by experimental error or by some unaccounted sorption mechanism. It can only be speculated that Cs can be sorbed at pH 4–6 on organic substances not removed from the surface by leaching of the clay during preparation of sample S(2). The effect of organic coatings was recently reported for Cs sorption on soil [29]. It was even found that organic matter coatings can have a larger effect on Cs sorption to clay minerals than mineral coatings and high-affinity sites can be blocked by organic substances. Cs interaction with humic acids was found to be negligible at higher ionic strength [5, 29].
The increase in NaNO3 concentration to 0.1 mol/L brings about a pronounced decrease in Cs sorption on both the clay samples and diminished scatter of sorption values. The decrease is due to competition of Na+ ions in the ion exchange of Cs. The exchange on the edge sites of the untreated clay is virtually eliminated and the exchange on the layer sites is strongly suppressed. If there is an unaccounted sorption mechanism at pH 4–6, it is also eliminated. Nevertheless, the modeling results suggest that edge sites exist on the pretreated clay which can exchange Cs at pH > 8.
The effect of Na+ ions on Cs sorption to clay S(2) is smaller, which can indicate the existence of two kinds of layer sites with different selectivity for Cs. It can be supposed that the sites of low selectivity are located mainly on the surface coatings, and therefore sodium ions strongly suppress Cs sorption on the untreated clay S(1). Removal of the coatings changes the effect of Na+ ions since the layer sites of higher selectivity become accessible. However, this explanation is not supported by the data on the effect of the ionic strength and coatings removal on the concentrations of layer and edge sites in the clay samples (Table 1) which can be due to the presence of organic substances in the coatings.
Description of sorption of Am and Pu and analysis of its mechanism is more difficult due to hydrolysis of both the elements (Fig. 3). Hydrolysis of trace elements often results in an increase of their physical adsorption and chemisorption as well as formation of their colloidal forms [30]. This may complicate application of sorption models based on simple sorption equations and is probably responsible for the imperfect fitting of experimental data shown in Figs. 4 and 5, particularly in the case of Pu and Am sorption on the clay. Nevertheless, the curves of solid speciation representing changes in the speciation (bonding) of the elements in the solids give at least a good picture of possible mechanisms of Am and Pu sorption. It can be seen that ionic strength changes do not affect solid speciation of Pu(IV), whereas speciation of Am(III) varies depending on ionic strength (Fig. 4). These variations can be attributed to the changes in the aqueous speciation.
The calculated solid speciation suggests that at low pH values (3.5–6) Pu is predominantly sorbed on all the studied solids by ion exchange on layer sites. At the higher pH values bonding on edge sites also takes place, as an exception is the clay. The small effect of the ionic strength on the sorption of Pu on the clay (Fig. 4) indicates that the ion exchange of Pu significantly differs from that of Cs and Am due to the exchange of hydrolyzed Pu species. From the comparison of the calculated and experimental data for the natural clay with those for the potential coating minerals it can be concluded that the studied coating minerals play a minor role in Pu sorption on the clay at the low (<4–5) and high (>8 for goethite) pH values. Probably other coating minerals present in the clay (e.g. ferrihydrite [23, 31]), and/or organic substances (unpublished data) could enhance Pu uptake to clay particles.
The modeling of Am sorption on all the solids resulted in a more complicated picture of sorption mechanisms (Figs. 4, 5). In general, ion exchange on layer sites predominates, with Am3+ and AmNO3 2+ sorbed in different proportions and pH ranges. The sorption of Am3+ is the highest at most pH values, except for Am sorption on the clay from 0.01 mol/L NaNO3. Formation of Am surface complexes on edge sites was calculated as significant only at the higher pH values: SOAmOH at pH 5–8 on magnetite and pH > 7 on hematite, SOAm(OH)2 at pH > 7 on hematite and pH 7–8 on the clay. These results indicate that ion exchange on layer sites of iron oxide coatings of the clay can play a certain role in Am sorption on the clay but the importance of the iron oxide coating sites is different. However, results of the modeling of Am and Pu sorption must be interpreted with caution, and further studies should be made to elucidate mechanisms of the sorption and the role of other coating components of clay not considered in this study.
Conclusions
Studies of pH dependence of Cs sorption in the presence and absence of clay coatings indicated that the coatings can enhance Cs sorption at the low ionic strength (0.01 mol/L NaNO3) and inhibit Cs sorption at the higher ionic strength (0.1 mol/L NaNO3. Two sorption mechanisms were assumed: at pH < 6, ion exchange on the layer sites and ion exchange on the edge sites at pH 7–10.
Pu sorption and speciation were not affected by changes in the ionic strength, whereas speciation of Am(III) varied depending on ionic strength. Comparison of the calculated and experimental data has shown that the studied coating minerals play a minor role in Pu sorption on the clay at the low (<4–5) and high (>8 for goethite) pH values. It is, however, possible that other coating minerals, e.g. ferrihydrite, and/or organic substances present in the clay enhance Pu uptake to clay particles.
The modeling of Am sorption has indicated that ion exchange on layer sites predominates, with Am3+ and AmNO3 2+ sorbed in different proportions and pH ranges. Formation of Am surface complexes on edge sites was calculated as significant only at the higher pH value.
References
Cornell RM (1993) J Radioanal Nucl Chem 171:483–500
Dumat C, Quiquampoix H, Stauton S (2000) Environ Sci Technol 34:2985–2989
Bostick BC, Vairavamurthy MA, Karthikeyan KG, Chorover J (2002) Environ Sci Technol 36:2670–2676
Steefel CI, Carroll S, Zhao P, Roberts S (2003) J Contam Hydrol 67:219–246
Bellenger JP, Staunton S (2008) J Environ Radioact 99:831–840
Perret D, Gaillard JF, Dominik J, Atteia O (2000) Environ Sci Technol 34:3540–3546
Singh BK, Jain A, Kumar S, Tomar BS, Tomar R, Manchanda VK, Ramanathan S (2009) J Contam Hydrol 106:144–149
Xu X, Kalinichev AG, Kirkpatrick (2006) J Geochim Cosmochim Acta 70:4319–4331
Choppin GR (2006) Mar Chem 99:83–92
Reiller PE, Evans NDM, Szabo G (2008) Radiochim Acta 96:345–358
Andre C, Choppin GR (2000) Radiochim Acta 88:613–616
Banik NL, Buda RA, Burger S, Kratz J, Trautmann N (2007) J Alloys Compd 444–445:522–525
Lujanienė G, Plukis A, Kimtys E, Remeikis V, Jankūnaitė D, Ogorodnikov BI (2002) J Radioanal Nucl Chem 251:59–68
Desideri D, Meli MA, Roselli C, Testa C, Degetto S (2001) J Radioanal Nucl Chem 248:727–733
Lusa M, Lehto J, Leskinen A, Jaakkola T (2009) J Environ Radioact 100:468–476
McCubbin D, Leonard KS, Young AK, Maher BA, Bennett S (2004) J Environ Radioact 77:111–131
Kenna TC (2009) J Environ Radioact 100:547–557
Mincher BJ, Fox RV, Cooper DG, Groenewold GS (2003) Radiochim Acta 7:397–402
Mehra OP, Jackson ML (1960) Clays Clay Miner 7:317–327
Schwertmann U, Cornell RM (1991) Iron oxides in the laboratory. VCH, Weinheim
Zhang GY, Peak D (2007) Geochm Cosmochim Acta 71:2158–2169
Raming TP, Winnubst AJA, van Kats CM, Philipse AP (2002) J Colloid Interface Sci 249:346–350
Lujanienė G, Šapolaitė J, Amulevičius AS, Mažeika K, Motiejūnas S (2006) Czech J Phys SD56:D103–D110
Lujanienė G, Motiejūnas S, Šapolaitė J (2007) J Radioanal Nucl Chem 274:345–353
Wanner H, Albinsson Y, Karnland O, Wieland E, Wersin P, Charlet L (1994) Radiochim Acta 66/67:157–162
EPA Report (1999) 402-R-99-004A:450
Filipska H, Stamberg K (2006) J Radioanal Nucl Chem 270:531–542
Štamberg K, Venkatesan KA, Vasudeva Rao PR (2003) Colloid Surf A Physicochem Eng Aspects 221:149–162
Celebi O, Kilikli A, Erten HN (2009) J Hazard Mat 168:695–703
Beneš P, Majer V (1980) Trace chemistry of aqueous solutions. Elsevier, Amsterdam
Stumpf S, Stumpf T, Dardenne K, Hennig Ch, Foerstendorf H, Klenze R, Fanghanel Th (2006) Environ Sci Technol 40:3522–3528
Acknowledgments
The research was supported by the Agency for Science, Innovation and Technology of the Republic of Lithuania (contract No. TAP-36/2010) and the Research council of Lithuania (contract No. TAP-54/2010) as well as by the Ministry of Education of the Czech Republic (contract No. MSM 6840770020). The financial support is greatly acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lujanienė, G., Beneš, P., Štamberg, K. et al. Effect of natural clay components on sorption of Cs, Pu and Am by the clay. J Radioanal Nucl Chem 286, 353–359 (2010). https://doi.org/10.1007/s10967-010-0726-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-010-0726-y