
Abstract
Extraction behaviour of actinides, lanthanides, fission products and structural elements has been studied with the two diglycolamide extractants, namely N,N,N′,N′-tetra-2-ethylhexyl diglycolamide (T2EHDGA) and N,N,N′,N′-tetraoctyl diglycolamide (TODGA). The acid extraction studies suggested that T2EHDGA (KH: 1.8) is less basic as compared to its linear homologue, TODGA (KH: 4.1). The distribution ratio of Am(III) by 0.1 M diglycolamides followed the order: TODGA > T2EHDGA. The number of ligand molecules present in the stoichiometry of the extracted species of Am(III) was found to be three and four for T2EHDGA and TODGA, respectively. Thermodynamics studies suggested that the extraction of Am(III) by both the extractants is exothermic in nature. The radiolytic stability of TODGA and T2EHDGA solutions in n-dodecane has been investigated. Due to lower distribution ratio of Am by T2EHDGA, 0.2 M of its solution has been used as compared to 0.1 M solution of TODGA. The distribution behaviour of various metal ions, viz. Am, Nd, Fe, Mo, Cr, Sr and Cs has been studied from nitric acid as well as from simulated high level waste solution.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Disposal of High Level Waste (HLW) solution generated during the reprocessing of the spent nuclear fuel is of great environmental concern. At present, the accepted approach for the management of HLW is to vitrify it in borosilicate glass matrices followed by interim storage. It is proposed to be disposed off eventually in deep geological repositories. Since the half lives of minor actinides (long lived isotopes of Np, Am and Cm) range between a few hundred to millions of years, the surveillance of high active waste for such a long period is debatable from economics as well as environmental safety considerations. Therefore, the strategy of P&T (Partitioning of long-lived radionuclides followed by Transmutation in high energy high flux reactors or accelerator driven subcritical systems) is being considered by several countries worldwide [1]. After partitioning of the actinides along with long lived fission products, the residual waste can be vitrified and buried in subsurface repositories at a much reduced risk and cost.
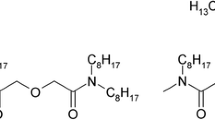
For the partitioning of minor actinides, several processes have been proposed, viz. TRUEX, DIAMEX and TRPO which employ octyl(phenyl))-N,N-diisobutyl carbamoyl methyl phosphine oxide (CMPO), N,N,N′,N′-dimethyldioctyl hexylethoxy malonamide (DMDOHEMA) and trialkyl phosphine oxide (TRPO), respectively as the extractants [2–4]. Each of these processes is associated with certain limitations. The main drawbacks of the TRUEX process are (a) the poor back extraction of Am(III) and Cm(III) at reduced acidity, and (b) interference due to solvent degradation products. On the other hand, TRPO solvent cannot be applied at 3–4 M HNO3 conditions generally encountered in HLW. Though the completely incinerable DMDOHEMA has been reported to be a promising candidate, it is a modest extractant of Am(III)/Cm(III) from HLW at acidity ≤3 M HNO3 [5, 6]. Recently, new class of diamide extractants, diglycolamides (containing ether linkage between two amide groups) have been the focus of many separation groups working on actinide partitioning. This class of extractants exhibit higher extraction of minor actinides as compared to malonamides due to their tridentate nature [7]. Amongst various derivatives of diglycolamides, N,N,N′,N′-tetraoctyl diglycolamide (TODGA, Fig. 1) has attracted attention and has been investigated intensively [7–12]. On the other hand, studies on its homologue, viz. N,N,N′,N′-tetra-2-ethylhexyl diglycolamide (T2EHDGA, Fig. 1) are scarce [13–15].

Structural formula of the diglycolamide extractants: R: n-octyl = TODGA; R: 2-ethylhexyl = T2EHDGA
Though TODGA [12] and T2EHDGA [13] have been studied on mixer-settler scale for actinide partitioning, the full potential of the latter in comparison to the former and relative merits of the two are not known. In the present paper, attempt has been made to compare the extraction properties of minor actinides with these two diglycolamide ligands. The extraction behaviour of Am(III) by the two extractants has been explained with the help of stoichiometry of the extracted species and various thermodynamics parameters. As TODGA and T2EHDGA have been proposed along with phase modifiers (viz. DHOA (N,N-dihexyl octanamide) and isodecanol, respectively) in mixer-settler studies, systematic studies for the extraction behaviour of various metal ions has been investigated from nitric acid medium as well as from Simulated High Level Waste (SHLW) solution using 0.1 M TODGA + 0.5 M DHOA and 0.2 M T2EHDGA + 30% (v/v) isodecanol in n-dodecane. The radiolytic stability of the above solvent mixtures has also been investigated.
Experimental
Reagents and radionuclides
TODGA and T2EHDGA were synthesized indigenously using a previously reported procedure [7]. The synthesized product was characterized using IR, PMR, HPLC and elemental analysis. Diluent, n-dodecane was procured from Lancaster and was used as received. All the other chemicals were of analytical reagent grade. 241Am was tested for its purity by α as well as γ spectrometry. Other radionuclides, viz. 85,89Sr, 59Fe, 137Cs, 51Cr and 99Mo were procured from Board of Radiation and Isotope Technology (BRIT), Mumbai, India and their purities were ascertained by gamma spectrometry using high purity germanium (HPGe) detector. The 147Nd tracer was obtained after irradiation of the natural Nd2O3 (specpure grade) in APSARA Reactor, Bhabha Atomic Research Centre at a neutron flux ~1012 neutrons/cm2/s. Pressurized Heavy Water Reactor Simulated High Level Waste (PHWR-SHLW) was supplied by Waste Management Division, BARC, Mumbai. The composition of this solution (Table 1) was equivalent to the generation of HLW from the fuel reprocessing of a typical PHWR of ~6500 MWD/T burn up and 3 years cooling and ~800 L of the waste solution per tonne of the spent fuel.
Distribution studies
For metal ions distribution studies, suitable volumes of aqueous phase (1 mL) containing varying concentrations of nitric acid (0.01–6 M) spiked with radiotracer were equilibrated with equal volume of organic phase in stoppered glass test tubes for 45 min in a thermostated water bath at 25 ± 1 °C. The two phases were then centrifuged and assayed radiometrically employing a well type NaI(Tl) gamma scintillation counter coupled with a multichannel analyzer. The distribution ratio of metal ion (DM) was calculated as the ratio of counts per minute in a given volume of organic phase to those in the identical volume of the aqueous phase. For acid distribution studies, equal volumes of organic and aqueous phases were equilibrated for 45 min followed by centrifugation. The acidity of the aqueous phase was subsequently determined by standard acid-base titration. The organic phase acidity was also determined by acid-base titration in 1:1 water : ethanol mixture previously neutralized to phenolphthalein indicator. All the experiments were carried out in triplicate and the accepted data were within the error limits of ±5%.
Results and discussion
Extraction of nitric acid
The diglycolamide ligands (A) interact with nitric acid as follows,
where the subscripts (aq.) and (org.) represent the aqueous and the organic phases, respectively, and KH is the acid uptake constant defined as,
Taking logarithm on both sides of Eq. 2 and rearranging,
where, [H+](org.) = [HNO3·nA](org.), and [A](org., free) = [A](initial) − [H+](org.). Here, [H+](aq.) was calculated from the titre value (T) and dissociation constant of HNO3 (Ka = 23.5) using the following equation,
where \( \left[ {{\text{H}}^{ + } } \right]_{{({\text{aq.}})}} \, = \, \left[ {{\text{NO}}_{3}^{ - } } \right]_{{({\text{aq.}})}} \) and T = {[H+](aq.) + [HNO3](aq.)} in aqueous phase (i.e. dissociated and un-dissociated HNO3). Therefore, Eq. 4 becomes,
From the Eq. 3 it is clear that a plot of {log[H+](org.) −2 log[H+](aq.)} against log[A](org., free) gives a straight line with slope n, i.e. the number of diglycolamide (DGA) molecules associated in the formation of DGA·HNO3 adduct and the intercept (log KH) gives the basicity (acid uptake constant) of the ligand. In view of the lack of data for the activities of HNO3 and free ligand in the organic phase, the concentration terms have been used and the two-phase equilibrium constant is referred to as “conditional acid uptake constant”. As a simplified case, for different concentrations of ligands equilibrated with 1 M HNO3, the plots for Eq. 3 were made assuming n = 1 (Fig. 2). From the intercept of the linear fits, the basicity (KH) of TODGA and T2EHDGA was calculated to be 4.1 ± 0.4 and 1.8 ± 0.3, respectively. The basicity of the T2EHDGA well agreed with that reported in the literature [15]. The results suggested that T2EHDGA is less basic as compared to the linear chain derivative of diglycolamides (TODGA). Though T2EHDGA is expected to be higher basic as compared to TODGA due to higher positive inductive effect (as branched alkyl groups show more +I effect), steric effect in the molecule seems to be dominant and hence reduce its acid complexation ability vis-a-vis TODGA.

Extraction of nitric acid by TODGA and T2EHDGA; diluent: n-dodecane; aqueous phase: 1 M HNO3; temperature: 25 °C
Extraction of americium
Kinetics of extraction
The rate of extraction of Am(III) by diglycolamide ligands was studied by equilibrating the organic phase and aqueous phase containing 241Am as tracer for different time intervals. The phase transfer kinetics was monitored in terms of fractional attainment of the equilibrium expressed as follows,
where, [Morg.]t and [Morg.]eq are the metal ions concentration in the organic phase at time ‘t’ and at equilibrium, respectively. The kinetics data were plotted in terms of (1-F) values as a function of equilibration time and are shown in Fig. 3. It is evident from the Figure that the equilibrium reached within 10 min for both the ligands. For T2EHDGA, however, the kinetics was found to be marginally slower as compared to TODGA. Further more, kinetics studies performed for a longer period (60 min) suggested that complexes of Am(III) and diglycolamide ligands are quite stable.

Kinetics of extraction of Am by TODGA and T2EHDGA; organic phase: 0.1 M TODGA or 0.1 M T2EHDGA; diluent: n-dodecane; aqueous phase: 1 M HNO3; temperature: 25 °C
Stoichiometry of the extracted species
In general, the two phase equilibrium representing the extraction of metal ions from nitric acid medium with neutral ligands like DGA (A) can be represented as,
The equilibrium constant for reaction (7) can be written as,
The experimentally determined distribution ratio (DM) is the ratio of the total concentration of the metal ion in the organic phase to the total concentration of the metal ion in the aqueous phase. Assuming that the (M(NO3)n·xA) is the only metal bearing species in the organic phase, the DM for the equilibrium reaction (7) can be expressed as,
Substituting the DM values in Eq. 8 and taking the logarithm of the rearranged equation,
At constant nitrate concentration (1 M HNO3), the DM values for Am(III) was measured at different ligand concentration. The stoichiometry of the extracted species was then obtained from the slope of the linear fits of the plot of log DM vs. log [A]. As shown in Fig. 4, the slope values obtained for T2EHDGA and TODGA were 3.02 ± 0.19 and 3.72 ± 0.29, respectively suggesting the stoichiometry of the extracted species as Am(NO3)3·3T2EHDGA and Am(NO3)3·4TODGA. Our results was consistent with the stoichiometry of the extracted of Am(III) with TODGA [7] and T2EHDGA [14] reported earlier. Higher solvation number for TODGA accounts for higher DAm values by TODGA as compared to T2EHDGA. It seems that TODGA molecules having linear alkyl chain may accommodate four ligand molecules easily around the metal ions as compared to T2EHDGA where steric hindrance restricts to only three ligand molecules with branched alkyl chains.

Extraction of Am(III) at varying concentration of diglycolamide; diluent: n-dodecane; aqueous phase: 1 M HNO3; temperature: 25 °C
Effect of nature of acid
The distribution studies of Am(III) from HCl, HNO3 and HClO4 medium suggested that for a given concentration of TODGA, the distribution values of Am(III) vary in the order: HCl < HNO3 < HClO4 [8]. It was of interest, therefore, to investigate the effect of nature of anion on distribution behaviour of Am(III) by branched alkyl derivative of diglycolamide (T2EHDGA). As shown in Fig. 5, the distribution behaviour of Am(III) by TODGA as well as by T2EHDGA were similar. The DAm increased sharply with nitric acid concentration up to 3 M beyond which a plateau was observed. On the other hand, lower DAm observed in HCl did not show much change upto 2 M HCl. However, there was a steep increase in DAm values beyond 4 M HCl. It was interesting to note that DAm values in perchloric acid medium were relatively larger and displayed little variation with aqueous phase acidity (0.01–6 M). High DAm in HClO4 was attributed to the ‘perchlorate effect’ where perchlorate ions promote the phase transfer of metallic cations by disrupting H2O structure in the aqueous phase [16]. It is worth mentioning that both the ligands, TODGA and T2EHDGA form third phase even when equilibrated with 6 M nitric acid solution. It has been found that the extraction of acid and water into the organic phase promotes the third phase formation [17]. In case of HCl, no third phase was observed for both the ligands as the extraction of water and acid is less in HCl [18]. For TODGA, the third phase was observed at 6 M HNO3, whereas for T2EHDGA it appeared even at 4 M HNO3. On the other hand, third phase formed more readily in HClO4 medium and 3 M HClO4 was sufficient to cause third phase formation. This behaviour may be attributed to the large amount of water and perchlorate ion extraction in the organic phase. Distribution ratios under such conditions were measured at enhanced organic to aqueous phase ratio.

Effect of nature of acid on the extraction of Am(III) by diglycolamide; organic phase: 0.1 M TODGA (open symbols) or 0.1 M T2EHDGA (closed symbols); diluent: n-dodecane; temperature: 25 °C
Thermodynamics of extraction
With the knowledge of stoichiometry of the extracted species, the extraction equilibria for Am(III) by TODGA T2EHDGA can be written as,
Assuming that the stoichiometry of the extracted species does not change in the temperature range employed in the present studies, the extraction constant of Am(III) for the above equilibrium reactions can be gives as follows [19],
The value of extraction constants (log Kex) for Am(III) with TODGA and T2EHDGA were calculated employing Eqs. 13 and 14, respectively. However to enable such calculations, it is essential to evaluate the value of \( \left( { 1+ \sum \beta_{\text{n}}^{\text{Am}} \left[ {{\text{NO}}_{ 3}^{ - } } \right]^{\text{n}} } \right) \). In the present work, this value of Am(III) was obtained from the literature [20]. Similarly, the value of free ligand concentration was determined from the basicity (KH) of the ligand. The Gibbs free energy change (∆G) associated with the extraction process can be calculated from the value of the extraction constant (Kex) employing the following equation,
The Gibbs free energy change (∆G) is correlated with the enthalpy change (∆H) and entropy change (∆S) of the system by Gibbs-Helmholtz equation as follows,
The thermodynamics parameters ∆H and ∆S were obtained from the linear fits of the plot of ∆G vs. T (Fig. 6). Detail studies on the extraction thermodynamics of actinides (Am, Pu and U) by TODGA has been reported earlier from our laboratory [21].

Variation of ΔG as a function of temperature for the extraction of Am(III); organic phase: 0.1 M DGA; diluent: n-dodecane; aqueous phase: 1 M HNO3
Table 2 lists the thermodynamic parameters for the extraction of Am(III) by TODGA and T2EHDGA. Negative enthalpy change suggests that the extraction process of Am(III) by both the extractants is exothermic in nature. More negative enthalpy change in case of TODGA is in expected line as the extracted species contains 4 TODGA molecules as compared to only 3 T2EHDGA molecules in the extracted species in the T2EHDGA extraction system. The data show that the Gibbs free energy change (−∆G) follows the order: TODGA > T2EHDGA. More negative ΔG value for TODGA suggests that the formation of tetra-solvated Am(III)-TODGA complex is thermodynamically more favoured as compared to tri-solvated complex with T2EHDGA. Similarly, higher exothermic enthalpy value (−∆H) for TODGA suggests the stronger binding of TODGA with Am(III) as compared to T2EHDGA. The over all enthalpy change depends on several contributing factors, such as, (i) dehydration of metal ions (∆H1); (ii) partitioning of ligand towards the aqueous phase(∆H2); (iii) formation of neutral extracted species (∆H3); and finally (iv) partitioning of the metal complex towards the organic phase (∆H4). The values of ∆H3 should be more exothermic for Am(III) as the formation of the tetra-solvated species in the organic phase is more exothermic as compared to tri-solvated species in case of T2EHDGA. Consequently, more exothermic ∆H was observed with TODGA Am(III) extraction. Extraction processes with both the DGA ligands are enthalpy driven whereas entropy factor counteracts the extraction. The negative entropy changes (−∆S) in the extraction of Am(III) could be due to the loss of translational and rotational entropy of DGA molecules upon complexation in the bulky extracted species, viz. Am(NO3)3.4TODGA and Am(NO3)3.3T2EHDGA. Less negative entropy change in case of TODGA suggests that more number of water molecules are released upon complexation with hydrated Am(III) from the inner co-ordination sphere. Consequently, it may be concluded that number of water molecules in the T2EHDGA-Am(NO3)3 complex is more as compared to TODGA-Am(NO3)3 complex.
Extraction of metal ions by diglycolamides (DGA)
Due to significantly lower DAm values by T2EHDGA vis-à-vis TODGA, 0.2 M of its solution has been recommended where 0.1 M TODGA is sufficient for extraction studies on trivalent actinides [8, 13]. However, due to low ligands concentrations, lower metal loading in the organic phase was observed. Therefore, suitable phase modifiers have been used along with these extractants to avoid third phase formation during actinide partitioning from HLW. In this context, 0.2 M T2EHDGA + 30% isodecanol in n-dodecane [13] and 0.1 M TODGA + 0.5 M DHOA (N,N-dihexyl octanamide) in n-dodecane [8] have been proposed. Further studies were, therefore, carried out using these solvents.
The extraction behaviour of various elements from nitric acid medium as well as from PHWR-SHLW by TODGA solvent has been reported earlier [8]. In this work, the extraction behaviour of various metal ions has been studied using T2EHDGA as the extractant and comparing the data obtained with TODGA. Figs. 7 and 8 show the distribution behaviour of different metal ions by T2EHDGA solvent, viz. Am(III), Nd(III), Tc(VII), Mo(VI), Cr(III), Fe(III), Sr(II) and Cs(I) from nitric acid solution as well as from PHWR-SHLW. The DM values for trivalent actinide and lanthanides are sufficiently large (at ~3 M HNO3) for the efficient extraction of these metal ions under HLW conditions. Negligible extraction of Cr, Fe and Cs was observed as DM values for these metal ions were <10−2 in the entire range of acidity investigated. The distribution of Cr was higher in SHLW at lower acidity which could be due to change in the oxidation state. The DSr value showed a maxima at ~3 M HNO3 where the value is 0.3. Interestingly, significant extraction of Mo and Tc was also observed at lower acidity (<0.1 M HNO3), which indicates that these metal ions may be extracted as protonated anionic species. In the presence of SHLW, the DM behavior of the above metal ions was similar to that in the nitric acid. However, the DM values were lower in the presence of SHLW as compared to pure tracers, which were attributed to the loading effect due to the co-extraction of other metal ions present in SHLW. The distribution ratio values of the metal ions from PHWR-SHLW of 3 M HNO3 are listed in Table 3. The table also compares the distribution values obtained with linear chain derivative of DGA, i.e. TODGA. It seems that the extraction behaviour of all the metal ions given in the table is same with both the extractants, though distribution ratio of trivalent actinides and lanthanides was significantly higher with TODGA. Under stripping conditions, DM values for Tc and Mo were significant for their interference during the stripping of the metal ions from the loaded organic phase. It is necessary, therefore, to choose the suitable feed condition where the extractions of these metal ions are suppressed.

Distribution behavior of metal ions by T2EHDGA; organic phase: 0.2 M T2EHDGA + 30% isodecanol; diluent: n-dodecane; aqueous phase: pure HNO3 (closed symbols) or PHWR-SHLW (open symbols); temperature: 25 °C

Distribution behavior of metal ions by T2EHDGA; organic phase: 0.2 M T2EHDGA + 30% isodecanol; diluent: n-dodecane; aqueous phase: pure HNO3 (closed symbols) or PHWR-SHLW (open symbols); temperature: 25 °C
Radiolytic stability of diglycolamides
In order to use the given extraction system for the separation of metal ions from HLW, it is essential to acquire the knowledge of radiation stability of the reagent. The radiation stability of TODGA has been studied and the G-value (number of molecules decreased after absorption of 100 eV of energy) was found to be 8.5 ± 0.6 [22]. It was found that the radiation stability of TODGA could be increased with the addition of benzene as well as DHOA. The main degradation products of TODGA are dioctylamine, dioctylacetamide, dioctylglycolamide and dioctylformamide because of the breaking of amide bonds as well as bonds in the vicinity of the ether oxygen. Excellent radiation stability of TODGA solvent (0.1 M TODGA + 0.5 M DHOA in n-dodecane) has been reported up to 500 kGy dose when the solvent was irradiated in contact with SHLW with 60Co source [10]. Modolo et al. [23] reported excellent radiolytic stability of 0.2 M TODGA + 0.5 M TBP in TPH upto 600 kGy even when the extractant was irradiated in contact with 3 M HNO3. In the present work, the radiolytic stability of T2EHDGA and TODGA the solvents was compared. The solvents were irradiated (in contact with equal volume of PHWR-SHLW in a glass stoppered bottle) under identical condition with gamma rays from 60Co source at an exposure dose of 1.8 KGy/h. Parts of the sample were withdrawn after suitable time interval to get an irradiation dose up to 1000 KGy. The resultant organic phase was employed for the measurement of distribution ratio of various metal ions from SHLW (extraction condition, Fig. 9) as well as from 0.01 M HNO3 (stripping condition, Fig. 10). The DM values for Am(III) and Nd(III) did not show any significant change upto 400 KGy in extraction cycle for both the solvents. However, the DM values for Am(III) and Nd(III) decreased after 400 KGy, suggesting radiolysis of the solvents. At the same time, the DM values of Am(III) and Nd(III) did not show any significant change under stripping condition, suggesting that the degraded products were not affecting the stripping of the metal ions. The radiation dose during actinide partitioning from actual HLW is likely to be in the range of few hundred kGy. Under such conditions, the radiation damage to the DGA solvents (TODGA and T2EHDGA) appears not to influence their performance in either extraction or stripping trivalent actinides and lanthanides. However, Tc shows little extraction with both the solvents and shows poor stripping at the same time.

Effect of gamma dose on the extraction of the metal ions from PHWR-SHLW by T2EHDGA (open symbol) and TODGA (closed symbol); organic phase: 0.2 M T2EHDGA + 30% isodecanol or 0.1 M TODGA + 0.5 M DHOA loaded with PHWR-SHLW; diluent: n-dodecane; temperature: 25 °C

Effect of gamma dose on the stripping of the metal ions by T2EHDGA (open symbol) and TODGA (closed symbol); organic phase: 0.2 M T2EHDGA + 30% isodecanol or 0.1 M TODGA + 0.5 M DHOA loaded with PHWR-SHLW; aqueous phase: 0.01 M HNO3; diluent: n-dodecane; temperature: 25 °C
Conclusions
Extraction behaviour minor actinides by TODGA and T2EHDGA solvents were studied. The basicity of T2EHDGA (KH = 1.8) was less than the linear chain derivative of DGA (TODGA, KH = 4.1). Slope analysis from nitric acid medium indicated that Am(III) is extracted as tri-solvated species by T2EHDGA and tetra-solvated species by TODGA. The extraction process of Am(III) by diglycolamide ligands was exothermic in nature. Extraction behaviour of other metal ions from nitric acid as well as from SHLW by DGA solvents (TODGA and T2EHDGA) followed a similar pattern. Though TODGA and T2EHDGA show similar extraction behaviour for actinide partitioning, TODGA appears to be more promising as it shows higher extraction of actinides / lanthanides even with lower concentration of ligand (0.1 M) as compared to T2EHDGA (0.2 M).
References
Baisden PA, Choppin GR (2007) Nuclear waste management and the nuclear fuel cycle. Radiochemistry and Nuclear Chemistry. In: Nagyl S (ed) Encyclopedia of life support systems (EOLSS). Eolss Publishers, Oxford
Nash KL, Madic C, Mathur JN, Lacquement J (2006) Actinide separation and technology. In: Morss LR, Edelstein NM, Fuger J, Katz JJ (eds) The chemistry of the actinide and transactinide elements, vol 4, 3rd edn. Springer, The Netherlands, pp 2622–2798
Naik PW, Dhami PS, Misra SK, Mathur JN (2003) Use of organophosphorus extractants impregnated on silica gel for extraction chromatographic separation of minor actinides from high level waste solution. J Radioanal Nucl Chem 257:327–332
Gupta B, Malik P, Deep A (2002) Extraction of uranium, thorium and lanthanides using Cyanex-923: their separation and recovery from monazite. J Radioanal Nucl Chem 251:451–456
Murillo MT, Espartero AG, Almaraz M, Quesada JS, Sugera M, Sanchez JC, Mendoza J, Prados P (2008) Radiochim Acta 241:241
Purroy DS, Baron P, Christiansen B, Glatz J-P, Madic C, Malmbeck R, Modolo G (2005) First demonstration of a centrifugal solvent extraction process for minor actinides from a concentrated spent fuel solution. Sep Purif Technol 45:157–162
Sasaki Y, Sugo Y, Suzuki S, Tachimori S (2001) The novel extractants, diglycolamides, for the extraction of lanthanides and actinides in HNO3-n-dodecane system. Solv Extr Ion Exch 19:91–103
Ansari SA, Pathak PN, Husain M, Parmar VS, Manchanda VK (2005) N,N,N′,N′-tetraoctyl diglycolamide (TODGA): a promising extractant for actinide-partitioning from high-level waste (HLW). Solv Extr Ion Exch 23:463–479
Sasaki Y, Choppin GR (2000) J Radioanal Nucl Chem 246:267
Ansari SA, Prabhu DR, Gujar RB, Kanekar AS, Murali MS, Natarajan V, Rajeswari S, Manivannan R, Antony MP, Srinivasan TG, Manchanda VK (2009) Counter-current extraction of uranium and lanthanides from simulated high level waste using N,N,N′N′-tetraoctyl diglycolamide. Sep Purif Technol 66:118–124
Hecke KV, Modolo G (2004) Separation of actinides from LLLW by extraction chromatography using novel DMDOHEMA and TODGA impregnated resins. J Radioanl Nucl Chem 261:269–275
Magnusson D, Christiansen B, Glatz J-P, Malmbeck R, Modolo G, Purroy DS, Sorel C (2009) Demonstration of a TODGA based extraction process for the partitioning of minor actinides from a PUREX raffinate Part III: Centrifugal contactor run using genuine fuel solution. Solv Extr Ion Exch 27:26–35
Manohar S, Sharma JN, Shah BV, Wattal PK (2007) Process development for bulk separation of trivalent actinides and lanthanides from radioactive high-level liquid waste. Nucl Sci Eng 156:96–102
Panja S, Ruhela R, Misra SK, Sharma JN, Tripathi SC, Dakshinamoorthy A (2008) A. Facilitated transport of Am(III) through a flat-sheet supported liquid membrane (FSSLM) containing tetra(2-ethylhexyl) diglycolamide (T2EHDGA) as carrier. J Membr Sci 325:158–165
Sharma JN, Ruhela R, Harindaran KN, Mishra SL, Suri AK (2008) Separation studies of uranium and thorium using tetra(2-ethylhexyl) diglycolamide (T2EHDGA) as an extractant. J Radioanal Nucl Chem 278:173–177
Tachimori S, Sasaki Y, Suzuki S (2002) Modification of TODGA-n-dodecane solvent with a monoamide for high loading of lanthanides(III) and actinides(III). Solv Extr Ion Exch 20:687–699
Roa PRV, Kolarik Z (1996) Solv Extr Ion Exch 14:955
Jensen MP, Yaita T, Chiarizia R (2007) Reverse-micelle formation in the partitioning of trivalent f-element cations by biphasic systems containing a tetraalkyl diglycolamide. Langmuir 23:4765–4774
Suresh G, Murali MS, Mathur JN (2003) Thermodynamics of extraction of Am(III) and Eu(III) from different anionic media with tri-n-octyl phosphine oxide. Radiochim Acta 91:127–130
Mathur JN, Nash KL (1998) Thermodynamics of extraction of Am(III) and Eu(III) from nitrate and thiocyanate media with octyl(phenyl)-N, N-diisobutyl carbamoyl methyl phosphine oxide. Solv Extr Ion Exch 16:1341
Ansari SA, Pathak PN, Husain M, Parmar VS, Manchanda VK (2006) Extraction of actinides using N,N,N′,N′-tetraoctyl diglycolamide (TODGA): a thermodynamic study. Radiochim Acta 94:307–312
Sugo Y, Sasaki Y, Tachimori S (2002) Studies on hydrolysis and radiolysis of N,N,N′,N′-tetraoctyl-3-oxapentane-1, 5-diamide. Radiochim Acta 90:161–165
Modolo G, Asp H, Schreinemachers C, Vijgen V (2007) Development of a TODGA based process for partitioning of actinides from a PUREX raffinate. Part I: Batch extraction optimization studies and stability tests. Solv Extr Ion Exch 25:703–721
Acknowledgement
Authors thank Dr. C.P. Kaushik of Waste management Division, BARC for providing PHWR-SHLW employed in the present work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gujar, R.B., Ansari, S.A., Murali, M.S. et al. Comparative evaluation of two substituted diglycolamide extractants for ‘actinide partitioning’. J Radioanal Nucl Chem 284, 377–385 (2010). https://doi.org/10.1007/s10967-010-0467-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-010-0467-y