
Abstract
A new extractant for the separation of actinide(III) and lanthanide(III) cations, bis(o-trifluoromethylphenyl) phosphinic acid (2) was synthesized. The synthetic route employed mirrors one that was employed to produce the sulfur containing analog bis(o-trifluoromethylphenyl) dithiophosphinic acid (1). Classic radiochemical methods and absorbance spectroscopy were used to study the coordination chemistry of the Am-dithiophosphinic acid and Am-phosphinic acid complexes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The minor actinides (MA) Am, Cm, and other transplutonium elements represent a significant, long-term hazard found in used nuclear fuel (UNF). The hazards of the MA elements are mainly related to the heat produced by their radioactive decay and the inherent radiotoxicity of the MA elements. The removal of these elements from UNF will reduce the overall hazards of the remaining material. Unfortunately, Am/Cm and Ln ions are most stable in the trivalent oxidation state and, as a result, have quite similar chemical behavior. Separation of the MA from the lanthanides is recognized as one of the most difficult challenges in separation science.
It has been argued that increased covalency in the interaction of An(III) ions with the soft donor atoms and/or changes in coordination geometries account for these effects [1–6]. There are many examples in the literature reporting the use of ligands incorporating either S [2, 7–9] or N [10–13] donor atoms for the separation of An(III) from Ln(III) species. The largest An(III)/Ln(III) selectivities reported utilized the dialkyldithiophosphinic acid, bis(2,4,4-dimethylpentyl)-dithiophosphinic acid, the active component of the commercial extractant Cyanex 301. Using Cyanex 301, Chen et al. [14] reported a separation factor of Am(III) from Eu(III) (SF = DAm/DEu) of SF ~ 4,700 in nitrate media.
Recent work at our laboratory [15, 16] has led to the development of a new synthetic pathway for the preparation of a more diverse series of dithiophosphinic acid (DPAH) extractants. One of the first DPAH extractants developed (ortho-trifluoromethylphenyl dithiophosphinic acid, referred to as 1), exhibits significantly better separation efficiency and extractant stability relative to commercially available examples [16].
In order to better understand this unusually effective separation behavior, the dioxo analog of SPA has been synthesized in our laboratory. In this paper, we describe the synthesis and elementary characterization of the dioxo analog, bis(o-trifluoromethylphenyl)phosphinic acid (2), and report a simple comparative extraction study that was conducted between the dithiophosphinic acid, 1 and dioxophosphinic acid, 2. The structures of the ligands used in this study are presented in Fig. 1.

Structures of compounds used in this study
Experimental
The following reagents were obtained from commercial sources as reagent grade or better and used without further purification: dichloro(diethylamino)phosphine [(Et2NPCl2] (Aldrich), phosphorus trichloride (Aldrich), magnesium shavings (Aldrich), o-trifluoromethylbromobenzene (Aldrich), m-trifluoromethylbromobenzene (Aldrich), lithium aluminum hydride (Aldrich), ammonium chloride (Aldrich), flower sulfur (Aldrich), anhydrous sodium sulfate (Aldrich), concentrated hydrochloric acid (trace metal grade, Fisher), and ammonium carbonate (Fisher). Solvents such as anhydrous diethyl ether, anhydrous tetrahydrofuran (THF), anhydrous toluene, toluene, petroleum ether, and hexanes were used as received (Aldrich). All reactions were carried out under a nitrogen atmosphere, unless otherwise indicated. The standard setup used the following: round bottom 3-neck flask, water-jacketed condenser, gas inlet adaptor and magnetic stir bar. Proton, 19F{1H}, 31P{1H} and 13C{1H} NMR spectra were recorded on a Bruker DMX 300WB spectrometer operating at 7.04 T: 300 MHz (1H), 282 MHz (19F), 121 MHz (31P) and 75 MHz (13C).
The synthesis of 1 is shown in Scheme 1 and described in detail in an earlier publication [15]. The synthesis of 2 uses similar phosphorus(III) chemistry while using two different ligands.

Synthesis of bis(o-trifluoromethylphenyl)-dithiophosphinic acid (1)
Synthesis of bis(o-trifluoromethylphenyl)phosphinic acid (2). The phosphorus(III) reactions (see Scheme 1) followed literature procedures. Anhydrous toluene (25 mL) was slowly added to the flask by syringe. Air was introduced into the flask by a pipette bubbler and the reaction was continuously stirred. This reaction was initially very vigorous (approx. 2 h) as the starting phosphine begins to react with oxygen. After this period, the reaction was slowly warmed to light reflux for 24 h. At this time, 31P NMR analysis showed ~50/50 mixture of (o-(CF3)C6H4)2POH and (o-(CF3)C6H4)2PO2H, with trace quantities of the starting phosphine. The toluene was removed at reduced pressure and the solids transferred to another 250 mL flask. Tetrahydrofuran (150 mL) was employed to dissolve the solids and an aqueous solution of sodium hydroxide (50 mL; 5.0 M NaOH) was added to the flask. This reaction mixture was vigorously stirred at ambient temperature for 40 h and the organic layer took on a slight red-orange color. The organic and aqueous phases spontaneously separated when the stirring was stopped. Using a 250 mL separatory funnel, the organic phase was separated, retained, and the removed at reduced pressure leaving an off-white solid. This solid was dissolved in diethyl ether (125 mL), placed in a 250 separatory funnel, and acidified by shaking with 4.0 M hydrochloric acid (75 mL). The organic layer was collected and the aqueous layer was washed twice with diethyl ether (25 mL). The ether extracts were combined, dried over anhydrous Na2SO4, and the ether was removed at reduced pressure. The solid product was recrystallized from diethyl ether/THF and gave prismatic crystals of the product, 2. Crystals were obtained in 25% yield; mp = 96–97 °C.
The trifluoromethylphenyl sulfone (FS-13) was obtained from Marshallton Research Laboratories Inc. (King, North Carolina) and used as received. All aqueous solutions were prepared using filtered, high resistivity water (Barnstead Nanopure). Distribution ratios for 241Am, 154Eu, and 22Na (DM = [M]org/[M]aq) were measured by equilibrium batch contacts between the organic and aqueous phases at an organic-to-aqueous phase ratio of unity (O/A = 1). In all experiments, the organic phase was pre-equilibrated by contacting three times with fresh aqueous phase containing the appropriate concentration of HNO3 and NaNO3, thus insuring all matrix components were present at equilibrium concentrations in the organic phase. For radiotracer experiments, the aqueous phase was of the appropriate HNO3, NaNO3 and stable Eu(NO3)3 concentrations spiked with the radionuclides of interest (241Am, 152,154Eu, and 22Na) in trace quantities (typically, less than 10−7 M each). The phases were mixed by vortex for 2 min which is sufficient time to ensure equilibrium was attained and separated by centrifugation. Each separated phase was sampled and the 241Am, 152,154Eu, and 22Na activity determined using gamma spectroscopy. The pcH of the aqueous phases were determined after the phases were separated using a pH meter and electrode calibrated at I = 1.0 M NaNO3.
The americium UV–Vis absorbance spectra were recorded using a micro-volume cell (Hellma) and an absorption spectrometer (Cary 50) equipped with a fiber optic interface. All experiments using macroscopic concentrations of americium were performed with 243Am, which was obtained from laboratory stocks. No Eu(NO3)3 was used in the macroscopic 243Am extraction experiments.
Results and discussion
Classic radioanalytical method of slope analysis is well known and well suited to the study of solvent extraction processes [17, 18]. The extractants used in this study behave as acidic liquid–liquid cation exchangers and the following chemical equilibrium applies:
where the subscripts ‘aq’ and ‘org’ refer to species in the aqueous or organic phases, respectively. This chemical reaction is described by the thermodynamic equilibrium constant, K, which is given by:
where the activity coefficients are ignored for simplicity.
By introducing the distribution ratio, D = [M]org/[M]aq and further simplifying Eq. 2 we arrive at the following equation which is the basis of classical slope analysis.
Using this expression, a set of experimentally determined distribution coefficients, which are measured under specified conditions, can be used to determine the equilibrium stoichiometry operative in the chemical reaction.
The acid dependence of the extraction of Am (circles), Eu (squares), and Na (diamonds) from nitric acid solutions by 1 and 2 dissolved in FS-13 are shown in Fig. 2. The slopes (m ~ 3.0) of the Am acid dependencies are consistent with an acidic, cation-exchange extraction mechanism in which three protons are exchanged to the aqueous phase for each metal cation complex formed in the organic phase. The significantly different slope observed for the Eu acid dependencies indicates the Eu-DPAH complex has a dramatically different stoichiometry or a fundamentally different interaction mode with these extractants. In fact, the DEu values are likely an “optimistic” estimate of very low Eu partitioning in this system. The lack of an acid dependence in the case of the sodium ion extraction is likely due to saturation which occurred during the pre-equilibration of the ligand solutions.

Acid dependence of the extraction of Am (circles), Eu (squares), and Na (diamonds) by 1 (left) and 2 (right). Organic phase: 0.1 M ligand in FS-13. Aqueous phase: 0.001 M Eu(NO3)3, varying [HNO3], M total 1.0 M NaNO3
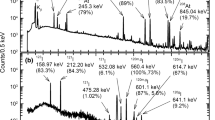
In order to explore the differences in the coordination chemistry of the dithio- and dioxo-phosphinic acids, extraction experiments were performed using macroscopic concentrations of Am(III) ion. In these high metal concentration experiments, 243Am was used without any addition of Eu(NO3)3 or other radiotracers. In order to minimize any variations in the pcH of the aqueous phases upon Am extraction, sulfanilic acid was employed as an aqueous phase buffer. The UV–Vis absorbance spectrum of the 4.0 × 10−4 M Am(III) solution used for these experiments is shown in Fig. 3. The absorbance spectrum exhibits the characteristic, sharp Am(III) band at ~503 nm and a much broader band at ~810 nm.

UV–Vis absorbance spectrum of 4.0 × 10−4 M Am(III) in 0.1 M sulfanilic acid, 0.9 M NaNO3, pcH = 2.0
The “macro” Am extraction experiments were performed as previously described and the absorbance spectra of the loaded organic phases were recorded. The UV–Vis absorbance spectra of the Am-dithiophosphinic acid complex dissolved in FS-13 and the Am-dioxophosphinic acid complex dissolved in FS-13 are shown in Fig. 4. The solid traces in Fig. 4 correspond to the loaded organic phase and the dashed traces correspond to the blank organic phase. The values of DAm for the extraction of Am by 1 or 2 in the presence of 0.1 M sulfanilic acid were also measured. For ligand 1, DAm = 0.4 and for ligand 2 DAm = 4. These distribution ratios indicate that approximately 20% of the available Am was extracted by 1, while 2 extracted approximately 80% of the available Am.

UV–Vis absorbance spectrum of a 0.1 M “1” dissolved in FS-13 and b 0.1 M “2” dissolved in FS-13. Prior to contact (dashed) and after contact (solid) with solution of 4.0 × 10−4M Am(III) in 0.1 M sulfanilic acid, 0.9 M NaNO3, pcH = 1.9
While the differences in the measured Am distribution ratios and acid dependencies (see Fig. 2) would seem to indicate that these two ligands behave differently, the observed UV–Vis absorbance spectra point to dramatically different Am coordination environments in the two ligand systems. In the case of the Am-dithiophosphinic acid (1) complex, an intense ligand to metal charge transfer (LMCT) band is observed in the UV–Vis spectrum as well as a sharp absorbance at ~515 nm which is attributable to an Am(III) metal-centered absorption. The observation of an Am(III) metal-centered absorption feature indicates that the Am-dithiophosphinic acid complex formed in the organic phase possesses a high symmetry. In the case of the UV–Vis absorbance spectra of the Am-dioxophosphinic acid complex, the lack of any significant absorption bands points to the metal being present in a completely different coordination environment of much lower symmetry.
Conclusions
A novel synthetic pathway was used to synthesize bis(o-trifluoromethylphenyl)-dithiophosphinic acid and bis-(trifluoromethylphenyl)phosphinic acid extractants. Radioanalytical methods were used to establish that these phosphinic acids behave as liquid-liquid cation exchangers. The results of slope analysis indicate that the Am and Eu phosphinic acid complexes have different stoichiometries. Absorption spectra recorded for the Am-dithiophosphinic acid complex and the Am-phosphinic acid complex indicate that the two complexes have coordination environments with widely differing symmetries. Work continues in our labs to better understand the behavior of these and other structurally similar DPAH extractants.
References
Diamond RM, Street K, Seaborg GT (1954) J Am Chem Soc 76:1461
Jensen MP, Bond AH (2002) J Am Chem Soc 124:9870
Mehdoui T, Berther JC, Thuery P, Ephritikhine M (2005) Dalton Trans 1263
Miguirditchian M, Guillaneus MD, Guillaumont D, Moisy P, Madic C, Jensen MP, Nash KL (2005) Inorg Chem 44:1404
Petit L, Adamo C, Maldivi P (2006) Inorg Chem 45:8517
Mathur JN, Murali MS, Nash KL (2001) Sol Extr Ion Exch 19:357
Modolo G, Nabet S (2005) Sol Extr Ion Exch 23:359
Zhu Y (1995) Radiochim Acta 68:95
Guoxin T, Yongjun Z, Jingming X, Ping Z, Tiandou H, Yaning X, Jing Z (2003) Inorg Chem 42:735
Giest A, Hill C, Modolo G, Foreman M, Weigl M, Gompper K, Hudson M (2006) Sol Extr Ion Exch 24:463
Jensen MP, Beitz JV, Rogers RD, Nash KL (2000) Dalton Trans 3058
Drew MGB, Iveson PB, Hudson MJ, Liljenzin JO, Spjuth L, Cordier PV, Enarsson A, Hill C, Madic C (2000) Dalton Trans 821
Kolarik Z, Mullich U, Gassner F (1999) Sol Extr Ion Exch 17:1155
Chen J, Zhu Y, Jiao R (1996) Sep Sci Technol 31:2723
Klaehn JR, Peterman DR, Tilloston RD, Luther TA, Harrup MK, Law JD, Daniels LM (2008) Inorg Chim Acta 361:2522
Peterman DR, Greenhalgh MR, Tillotson RD, Klaehn JR, Harrup MK,. Luther TA, Daniels LM (2008) In: Proceedings of ISEC 2008 International Solvent Extraction Conference, Canadian Institute of Mining, Metallurgy and Petroleum, Montreal, Quebec, Canada, p 1427
Baes CF, Mcdowell WJ, Bryan SA (1987) Sol Extr Ion Exch 5:1
Otu EO, Chiarizia R, Rickert PG, Nash KL (2002) Sol Extr Ion Exch 20:607
Acknowledgments
This work was supported by the United States Department of Energy and the Laboratory Directed Research and Development (LDRD) program at the Idaho National Laboratory (INL) through contract DE-AC07-05ID14517.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peterman, D.R., Martin, L.R., Klaehn, J.R. et al. Selective separation of minor actinides and lanthanides using aromatic dithiophosphinic and phosphinic acid derivatives. J Radioanal Nucl Chem 282, 527–531 (2009). https://doi.org/10.1007/s10967-009-0288-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-009-0288-z