
Abstract
The stabilization of natural rubber (NR) was studied using four NR samples, namely, whole natural rubber (WNR), deproteinized natural rubber (DPNR), acetone extracted natural rubber (ANR) and purified natural rubber (PNR). Proteins and lipids, which are non-rubber components in NR, significantly affect storage hardening via gel formation. The main function of the applied viscosity stabilizer was to perturb the dynamics of the NR structure, or alternatively, the formation of gel networks of the rubber chains with lipids and proteins, respectively. The Mooney viscosity and molecular weight of NR samples with the viscosity stabilizer added were found to be almost constant after 12 weeks. Moreover, the NR samples with the viscosity stabilizer required a longer curing time, presumably due to less gel network formation and the conventional packing of linear rubber chains. The results from crosslinking density and molecular weight measurements revealed that the viscosity stabilizer also had a role in stabilizing crosslinks between the chains and preventing the reversion to NR. The new approach of on structure-property relationships of stabilized natural rubbers has been proposed.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Natural rubber (NR) has many attractive properties such as high green strength, high tensile and tear strengths, good crack growth resistance, minimal heat build-up and good processability [1, 2]. Moreover, it is a natural and renewable polymer with outstanding properties contrary to synthetic rubbers. Therefore, NR has been used in many applications, for example, surgical gloves, condoms, thin films, colloids, liquid fuels, adhesive automotive parts, medical products and has had significant use in the tire industry [3,4,5,6,7]. However, one of the major problems during prolonged storage of NR is an increase in viscosity, called storage hardening. The occurrence of storage hardening possibly happens from crosslinking and chain-scission reactions between some abnormal groups such as epoxide, aldehyde and lactone [8,9,10] in the isoprene chain with amino acids and proteins contained in NR. Moreover, the gel fraction is generated through reactions between proteins and phospholipids existing in NR (about 5–6%) and the functional terminal groups of the NR molecules [10, 11]. From this effect, the viscosity of NR increases during storage. A molecular chain of NR is composed of two or three units of trans-1,4-polyisoprene and a long sequence of cis-1,4-polyisoprene. At the initiating terminal, named the ω-terminal, the NR molecule associates with proteins, while the chain end, called the α-terminal, associates with phospholipids. The NR molecules can form gel fractions through reactions between functional terminal groups at the end of the NR molecules and proteins at the ω-terminal or phospholipids at the α-terminal [10,11,12].
High viscosity NR requires higher energy and time to masticate before using it, which can also cause mechanical deterioration of a mixing machine’s components. In order to prevent gel formation and maintain a constant viscosity of NR, a viscosity stabilizer was applied. Hydroxylamine hydrochloride is typically used as a viscosity stabilizer. There have been several studies of the storage hardening effect on NR properties [13, 14]. Surprisingly, only a few investigations were about a viscosity stabilizer in NR, particularly on the dynamic structure of NR affected by the proteins and lipids in NR. NRs containing different contents of non-rubber constituents (WNR, DPNR, ANR and PNR) were prepared in this study from Hevea latex, according to a well-defined method [12]. Herein, the aim of this work is to study the effect of non-rubber components, especially proteins and lipids, on the chemical and physical properties of viscosity stabilized NR (uncrosslinking) at various storage times and also crosslinked NR. New information from this work may benefit current natural rubber applications as a model system.
Experimental
Preparation of NR samples
Fresh natural rubber latex is from Hevea brasiliensis clone RRIM 600. Latex is preserved by adding 0.2% v/v of 30% ammonium hydroxide (low ammonium content). Four groups of rubber samples were prepared from the latex according to the previous procedure [12] as follows.
Whole Natural Rubber (WNR) was prepared by casting fresh natural rubber latex on glass plates and air-drying for a day at room temperature. The rubber samples were then put in an oven at 50 °C for 24 h.
Deproteinized Natural Rubber (DPNR) was prepared from latex centrifugation at 10,000 rounds per minute (rpm) for 30 min. The cream fraction was collected and washed in sodiumdodecylsulfate (SDS; 1% w/v) and deionized water, respectively. After protein removal, the rubber was casted into a thin film and dried at 50 °C for 24 h in an oven.
Acetone extracted Natural Rubber (ANR) was prepared from WNR by acetone extraction in a Soxhlet apparatus for 16 h. The extracted rubber was dried at 50 °C for 24 h.
Purified Natural Rubber (PNR) was prepared from DPNR and then by acetone extraction in the same way as ANR, respectively.
Preparation of stabilized and unstabilized NR samples of uncrosslinked system
Unstabilized NR samples: two hundred grams of rubber samples (WNR, DPNR, ANR and PNR) were individually masticated using a two-roll mill. The front-rotor speed was 18 rpm and the back-rotor speed was 20 rpm. The mastication for each sample was operated at 70 °C for 6 min.
Stabilized NR samples: all the NR samples were masticated at 70 °C for 3 min (the same as the unstabilized system), subsequently 0.4 g of hydroxylamine hydrochloride was added. The mastication was continued for 3 min. The rubber samples were kept for 12 weeks at room temperature. Then, the rubber samples at 0 and 12 weeks were characterized respectively.
Preparation of stabilized and unstabilized NR samples of crosslinked system
Unstabilized NR samples: WNR and PNR (200 g each) were separately masticated using a two-roll mill with a front-rotor speed of 18 rpm and a back-rotor speed of 20 rpm at 70 °C for 5 min. Then, 6 g of ZnO mixed with 2 g of stearic acid were added and mastication continued for 2 min. N-tert-butyl-2-benzothiazyl sulphenamide, TBBS (4 g) was added and continuously masticated for 1 min. Finally, 3 g of sulphur was added and masticated for 2 min. The rubber samples were further masticated for 10 min for good dispersion of chemicals in the samples.
Stabilized NR samples: WNR and PNR (200 g each) were separately masticated using a two-roll mill with a front-rotor speed of 18 rpm and a back-rotor speed of 20 rpm at 70 °C for 4 min. Then, 0.4 g of hydroxylamine hydrochloride (0.2 phr) was added and mastication continued for 1 min. Then, ZnO, steric acid, TBBS and sulphur were added and masticated in the same way as the unstabilized system. The total mastication time was the same as that of the unstabilized system.
Characterization
Nitrogen content
The nitrogen content of the NR samples (WNR and PNR) was determined using the Kjeldahl method [2]. Dried rubber sheets were cut into 0.5 g pieces and placed in a Kjeldahl flask. Then, 0.8 g of the catalyst mixture (K2SO4:CuSO4•5H2O in 7:0.8 by mass) was added, followed by 15 ml of concentrated sulfuric acid. The mixture was boiled gently in the digestion unit at 420 °C until the solution became colourless. The digested solution was cooled to room temperature, and then transferred into a distillatory using 25 ml of 4% H3BO3 as the receiving solution. The distillation continued until 200 ml of distillate was collected. Thereafter, the distillate was titrated with 0.01 M HCl. A blank was determined by adding all the reagents but omitting the samples. The total nitrogen content was calculated as follows:
where B is ml of HCl required for titration of the receiving flask, C is ml of HCl required for titration of the blank, W is mass of sample (g) and N is concentration of HCl (N).
Lipid content
The lipid content of the NR samples was assessed through extraction methods [2]. For this, first, small pieces of rubber (2.8 g) were added to a vibratory miller machine with liquid nitrogen for grinding. The ground, frozen rubber was then extracted in chloroform: methanol (2:1) with agitation at 150 rpm for 6 h. The extracted rubber was filtered and rinsed with the extracting solvent. The extracted rubber was then dried at 100 °C for 1 h and then weighed. The filtrate was evaporated using a rotary evaporator. The corrected residue was re-dissolved with 1 ml chloroform:methanol (2:1). The total extracted solution was then washed with 1 ml of 0.9% NaCl solution in order to separate water soluble components from lipids. The lipid component, separated at the bottom layer, was collected and the solvent was evaporated.
Gel content
The gel content was determined by dissolving small pieces of rubber in toluene (0.1%, w/v) and the solution was kept in the dark at room temperature, without shaking or stirring, for one week. The solution was centrifuged at 10000 cycles/min for 45 min to separate the gel fraction. The gel was coagulated with methanol and dried under vacuum at room temperature [12].
FTIR spectra
The functional groups of proteins and lipids in NR were examined by Attenuated Total Reflection Fourier Transform Infrared, ATR-FTIR (BRUKER, EQUINOX 55, USA) using a Ge crystal probe. The spectra were recorded by 32 scan summations and a 4 cm−1 resolution.
Mooney viscosity
A Mooney viscometer consists of a rotating disc imbedded in a rubber specimen contained within a sealed, pressurized and heated cavity. The rubber sample is heated for 1 min before starting the motor. After that, the rubber sample is continuously measured for 4 min by the torque required to keep the rotor rotating at a constant rate as a function of time for reading the Mooney viscosity which is recorded as torque in N·m.
Molecular weight (MW) and molecular weight distribution (MWD)
The MW and MWD of the rubbers were determined by gel-permeation chromatography (GPC), using tetrahydrofuran (THF) as an eluent at a flow rate of 1 ml/min. Commercial standard polystyrenes were used for calibration.
Rubber processing analysis (RPA)
The processibility under strain sweep modes of the rubber samples was investigated by a rubber processing analyser (RPA 2000, Monsanto, USA). The strain sweep mode was in the range of 0.5–100%. The process was carried out at 100 °C and 1 Hz.
Optimum curing time
The optimum curing time of the compounded rubber samples was determined using a moving die rheometer (MDR) at 150 °C.
Crosslinking density and molar mass between crosslinks
The crosslinking density of rubber networks was measured by the swelling method. A small piece of the crosslinked rubber was soaked in toluene until the swelling equilibrium was reached. The number of effective network chains per unit volume of rubber (ν) was calculated, according to the Flory–Rehner equation [8]:
where Vs is the molar volume of toluene (106.9 cm3/mol), Vr is volume fraction of rubber in the swollen network, χ is the solubility parameter between polymer and solvent (0.43 + 0.05Vr). The samples were removed from the solvent every day for 7 days, weighed, dried for 1 week at 60–70 °C and for 1 day under vacuum and weighed again. The effective network concentration (ν) is related to the molecular weight between crosslinks Mc according to ν = ρr/Mc, where ρr is the density of rubber (NR = 0.93 g/cm3).
Tensile test
The rubber samples were cut into a dumbbell shape using a hydraulic cutting machine in accordance with the ISO 37 (die type 2) test method. The rate of testing crosshead speed was 500 mm/min at room temperature. The applied load cell was 1 kN. The tensile test was repeatedly run 5 times in total.
Results and discussion
Chemical characterization of NR samples
The NR samples were prepared from fresh NR latex – Hevea brasiliensis clone RRIM 600 – which was stabilized with high ammonium content. In the present work, proteins and lipids are two major components to be studied for their effect on dynamic viscosity of NR. Therefore, four NR samples with different non-rubber components were prepared as shown in Fig. 1. WNR was prepared from NR latex as received, so it contains both proteins and lipids. DPNR, where its protein was removed by centrifugation, contained lipids in major. While ANR remained proteins in major because lipids were removed by acetone extraction. Lastly, PNR was mostly purified by the removal of proteins and lipids. All NR samples were characterized for their nitrogen and lipid contents.

Schematic models of the natural rubber samples: Whole Natural Rubber (WNR), Deproteinized Natural Rubber (DPNR), Acetone extracted Natural Rubber (ANR) and Purified Natural Rubber (PNR)
Proteins in NR, determined by the Kjeldahl method for nitrogen content, involve both proteins in the serum phase and those on the surface of latex particles which is the protein on the isoprene chains at the ω-terminal. The lipid content of the NR samples which are related to phospholipids was determined by using chloroform: methanol extraction. The results of the nitrogen and lipid contents are shown in Table 1. DPNR and PNR had a very low nitrogen content, the proteins were soluble in SDS during the centrifugation process [2, 15]. For ANR which was prepared from WNR, the same nitrogen content as WNR was observed. The results suggested that centrifugation in the presence of a surfactant could be an effective method to reduce protein in the rubber samples. Concerning the lipid content analysis, the decrease of %lipid of DPNR compared to WNR corresponded to the removal of some lipids during the centrifugation process [16]. After acetone extraction, the lipid was removed from WNR and only 0.73% of the lipid remained in the ANR sample. Likewise, the lipid was extracted from DPNR to yield PNR with lower lipid content.
Stabilized and unstabilized NR samples of an uncrosslinked system
Each of the NR samples was masticated to prepare the NR samples with and without the viscosity stabilizer. After that, the masticated NR samples were stored at 0 and 12 weeks in order to study the dynamic properties of different NR samples at various storage times.
The chemical compositions of the NR samples, especially proteins and lipids, were determined by ATR-FTIR. The FTIR spectra of the NR samples without the viscosity stabilizer (unstabilized NR samples) at 3600–2600 cm−1 and 1800–1600 cm−1 are shown in Fig. 2, respectively. At week 0, WNR showed peaks of the amine group at 3280 cm−1, the fatty acid ester group at 1740 cm−1, the aldehyde group at 1710 cm−1 and the amide I group at 1660–1630 cm−1. In the case of DPNR, the peak of the amine group at 3280 cm−1 was not observed (Fig. 2a) and the intensity of the amide I group at 1660–1630 cm−1 decreased indicating that proteins were mostly removed from the NR sample. Lipids remaining in the DPNR sample were confirmed by peaks of the fatty acid ester and aldehyde groups at 1740 and 1710 cm−1, respectively (Fig. 2c). As expected, ANR samples gave the peaks of fatty acid ester and aldehyde groups at lower intensity than those of WNR since lipids were removed by acetone extraction. PNR was prepared from DPNR by further removing the lipid component, therefore, the peak of the amine group remained similar to that observed in DPNR. However, the peaks representing the lipid content were lower. PNR did not have peaks of the amine group at 3280 cm−1, the fatty acid ester group at 1740 cm−1, the aldehyde group at 1710 cm−1 and the intensity of the amide I peak at 1660–1630 cm−1 was lower than those of WNR. The results are in good agreement with the previous study [17]. After the NR samples were stored for 12 weeks, the peak intensity of the N-H group (Fig. 2b) and amide I (Fig. 2d) increased due to network formation, especially from proteins at the ω-terminal of the rubber chains. The higher intensity of carbonyl groups at 1740 and 1710 cm−1 after prolong storage demonstrated that gel formation also occurred from the lipid components.

FTIR spectra of NR samples at; a) 0 and b) 12 weeks without a viscosity stabilizer at 3600–2600 cm−1; c) 0 and d) 12 weeks without a viscosity stabilizer at 1800–1600 cm−1
The FTIR spectra of NR samples with a viscosity stabilizer (stabilized NR samples) are shown in Fig. 3. At 0 week, the samples gave similar results as those without a viscosity stabilizer. After 12 weeks, there was almost no change in the FTIR spectra except for the PNR sample. Generally, the viscosity stabilizer helps decrease storage hardening influencing from protein and lipid components in the samples. However, the higher intensity of the amide I peak in the case of PNR samples after 12 weeks was unexpected. This was presumably due to the excess viscosity stabilizer on the PNR surface which can be detected by FTIR around the amide I peak. The evidence of the excess viscosity stabilizer was brown spots in the PNR samples after 12 weeks as shown in Fig. 4.

FTIR spectra of NR samples at; a) 0 and b) 12 weeks with a viscosity stabilizer at 1800–1600 cm−1

Optical images of PNR with a viscosity stabilizer; left) 0 weeks and right) 12 weeks with samples size 2 cm by 2 cm
The gel content indicates the amount of network structure in NR which is non-soluble in solvent. Proteins and lipids play an important role in gel formation [12, 18]. The gel fraction was generated through the branch-point at the ω-terminal which is associated with proteins. The branch-point at the α-terminal was also formed by phospholipids. The gel content of the NR samples reported in Table 2 indicated that the proteins and lipids had an effect on the amount of gel in the samples. The WNR samples gave the highest gel content because of the high amount of lipids and proteins which can form gel networks at the α- and ω-terminals, respectively [19]. In contrast, the PNR samples gave the lowest gel content (12.1%) as both the proteins and lipids were mostly removed. The gel content of ANR and DPNR were lower than that of WNR. The gel networks in the ANR and DPNR samples could be generated mainly from the remaining proteins and lipids, respectively. The results are in good agreement with previous work [12]. The branch-point at the ω-terminal containing protein was decomposed by centrifugation with a surfactant [15]. However, DPNR had almost the same gel content as ANR. The gel formation in NR was therefore dependent on the protein and lipid fraction. The presumed structure of the gel fraction in the NR samples after purification has already been mentioned in Fig. 1.
The gel content also increased as the storage time increased. After 12 weeks, all NR samples had a higher gel content resulting from gel formation during prolonged storage. The increase of gel content in WNR, DPNR and ANR indicated that proteins and lipids are the major component in the formation of the gel fraction in stored NR samples. However, the increase of gel content observed in PNR was because of the small amount of proteins and lipids remaining in the sample.
Gel formation could cause storage hardening in NR. The effect of proteins and lipids on gel formation was less pronounced when the viscosity stabilizer, hydroxylamine hydrochloride, was applied. All NR samples with the viscosity stabilizer at 0 weeks exhibited lower gel content than those without. Thus, the important role of a viscosity stabilizer in the NR samples was to perturb the dynamic network formation at the end of rubber chains via proteins and lipids. Although, the gel content was higher after 12 weeks, the amount increased less obviously than the sample without a viscosity stabilizer. It is worth noting that storage hardening can be observed even in the constant viscosity rubber samples after prolonged storage [19].
Mooney viscosity indicates the viscosity of NR which depends on the macrostructure or the amount of gel fraction. The results of the Mooney viscosity of the sample at various storage times are shown in Table 2. At 0 weeks, WNR showed the highest Mooney value because the gel content was higher. DPNR and ANR almost had the same Mooney viscosity value, but were lower than WNR. PNR had the lowest viscosity value because proteins and lipids, which can cause gel formation, were removed indicating a smaller gel fraction in the sample. This result corresponds to the gel content. Considering the effect of the viscosity stabilizer, all NR samples with the viscosity stabilizer had lower Mooney viscosity than those without. After 12 weeks, the stored NR samples without viscosity stabilizer showed obviously higher Mooney viscosity than at 0 weeks. On the contrary, those with a viscosity stabilizer showed slightly or almost constant viscosity after long storage. This indicates that the viscosity stabilizer can inhibit dynamic Mooney viscosity in NR.
Generally, NR is an elastic polymer which has a high molecular weight in the range of 1.2 × 105 to 2.0 × 106 g/mol. The distribution of the NR molecular weight is well known as a bimodal molecular weight distribution (MWD), where the low molecular weight fraction is from 1.0 × 105 to 2.5 × 105 and the high molecular weight one is from 1.0 × 106 to 2.5 × 106 g/mol [19]. The molecular weight distribution of NR samples without a stabilizer at 0 and 12 weeks is shown in Fig. 5. At 0 weeks, the MWD of all NR samples were bimodal (Fig. 5a). For WNR, the high MW fraction was dominant. In contrast, the bimodal MWD of PNR showed a higher fraction at low MW. The difference in MWD of these NR samples was because of the difference in gel content. Both proteins and lipids in WNR could form gel networks resulting in a greater proportion of the high molecular weight fraction. On the other hand, PNR with a small amount of non-rubber components would be able to form less gel content. The molecular chains of PNR were mostly presented as linear chains which were a low molecular weight fraction in the distribution. In the case of the ANR and DPNR samples, their MWD was observed to be bimodal where the high molecular weight fraction was higher than PNR. This can be explained from the higher amount of proteins and lipids that remained in ANR and DPNR samples, respectively. The non-rubber components can cause gel formation which made ANR and DPNR have more branch or network structure than PNR. Thus, their high molecular weight fraction was higher than the low one.

Molecular weight distribution of rubber samples without a viscosity stabilizer; a) 0 weeks and b) 12 weeks
In Table 2, WNR had a higher Mw than those of purified NR because their protein and lipid content was higher. PNR had the lowest MW because most proteins and lipids were removed leading to less gel network formation. DPNR unexpectedly exhibited a little higher molecular weight than WNR which cannot be explained at present. However, the results are similar to a previous study [12]. The effect of storage time on the molecular weight and MWD of NR samples was determined at 12 weeks. The results showed that the MWD at 12 weeks of all NR samples was unimodal. The low molecular weight fraction almost disappeared and the peak observed was the high MW fraction. It was found that the Mw of WNR increased significantly with storage time because of the occurrence of storage hardening. However, there was no evidence found for other purified NR samples. For the NR samples with the viscosity stabilizer, the molecular weights observed at 0 and 12 weeks were very close. Thus, the storage time did not significantly affect the molecular weight of the stabilized NR samples. The viscosity stabilizer functioned as a dynamic network inhibitor between the rubber chain and non-rubber components.
Regarding the dynamic mechanical properties of uncrosslinked rubbers, the relationship between storage modulus (G’) and strain of rubber samples without the viscosity stabilizer at 0 and 12 weeks is shown in Fig. 6. The results showed that WNR possessed the highest G’ (highest rigidity), whereas PNR had the lowest G’. PNR had a short molecular chain because proteins and lipids at terminal ends were almost removed resulting in less gel and network structure formation in PNR. WNR had a higher gel fraction, molecular weight and strain-induced crystallization than the PNR samples [20, 21]. As a result, PNR had lower rigidity than WNR. At 12 weeks, the results were similar to those at 0 weeks, except for ANR, where G’ decreased. This may be because the lower lipid content in ANR affected the decrease of the low temperature crystallization in ANR [16]. Free fatty acids played an important role to accelerate the crystallization of natural rubber. The removal of free fatty acids from natural rubber resulted in a slower rate of crystallization, leading to a decrease in the strength of ANR.

Relationship between loss modulus (G’) and the strain of NR samples without a viscosity stabilizer at; a) 0 weeks and b) 12 weeks
Tan delta is the ratio of the loss modulus (G”) and storage modulus (G’). A high tan delta value means the rubber samples have lost more energy or have higher hysteresis. At 0 weeks, PNR (Fig. 7a) had the highest tan delta because proteins, fatty acids and phospholipids were mostly removed resulting in shorter molecular chains of PNR which could easily be moved, generating more dissipation of energy. WNR which had more network structure and longer molecular chains, generated less molecular friction than PNR. So, PNR has higher hysteresis than WNR. For tan delta of NR samples with the viscosity stabilizer at 0 weeks (Fig. 7b), they were similar to those without. After 12 weeks, the NR samples with and without the viscosity stabilizer illustrated the same tendency (data not shown here).

Relationship between tan delta and the strain of rubber samples at 0 weeks; a) without a viscosity stabilizer and b) with a viscosity stabilizer
A viscosity stabilizer affected the gel formation of NR by its interaction with proteins and lipids at the ω- and α-terminals, respectively (Fig. 8). We had an assumption that the interaction between non-rubber components and the viscosity stabilizer could be hydrogen bonding. This can be explained by the results of ATR-FTIR of uncrosslinked rubber. If the viscosity stabilizer covalently bonded with non-rubber components, the different functional group peaks would be observed as we compared the observed samples with and without a viscosity stabilizer. Thus, it is reasonable to conclude that a viscosity stabilizer interacted with proteins and lipids at the terminal ends via hydrogen bonding.

A viscosity stabilizer inhibited dynamic structure network of NR by its interaction with proteins and lipids at the terminal ends, respectively. The interaction between non-rubber components (proteins and lipids) and a viscosity stabilizer could be hydrogen bonding
Stabilized and unstabilized NR samples of a crosslinked system
In this part, the effect of the viscosity stabilization of vulcanized NR was studied. The samples in this study, WNR and PNR, were vulcanized by the sulphur system. Two types of rubber compounds were prepared, viz. the rubber compound with and without the viscosity stabilizer.
The optimum curing time of the rubber compounds was determined using the moving die rheometer at 150 °C. The results of curing time at t90 are reported in Table 3. After that, the rubber compounds were cured by using a vulcanization moulding machine at 150 °C according to their optimum curing time. The curing time of PNR was longer than WNR because both proteins and lipids were almost removed from PNR, especially proteins which act as a natural accelerator in the vulcanization of NR [21]. Based on the literature review, phospholipids also played an important role in the vulcanization reaction [23]. This can be stated that non-rubber components affect the curing time of NR vulcanization. Therefore, the crosslinking reaction in PNR was slower than in WNR. Regarding the effect of a viscosity stabilizer, we proposed that the viscosity stabilizer may interact with proteins and lipids at the chain ends of NR. Thus, the viscosity stabilizer could interrupt gel formation or the network structure in NR. Therefore, the samples with viscosity stabilizer were composed of more linear chains than those without. Time used for crosslinking the rubber chains with sulphur atoms would be longer than those without.
To study the effect of the viscosity stabilizer on a crosslinked rubber, the rubber compounds were cured according to their optimum curing time at t90, 2 × t90 and 3 × t90. The WNR samples were cured at 4, 8 and 12 min and the PNR samples were cured at 16, 32 and 48 min.
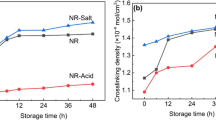
Focussing on the crosslinking density (ν) of NR samples, crosslinking density indicates a crosslinked network structure in the vulcanized NR (Fig. 9a). WNR had higher crosslinking density than PNR because WNR contained a greater gel content resulting in a higher molecular weight. Considering the effect of the viscosity stabilizer at 4 (t90) and 8 min (2 × t90), we found that WNR with and without the viscosity stabilizer at curing times of 4 and 8 min had similar ν values, but at 12 min (3 × t90) the value of WNR without the viscosity stabilizer slightly decreased. WNR without the viscosity stabilizer might degrade due to the reversion. WNR with the viscosity stabilizer had a constant ν value for all curing times. From this result, we can explain that the network structure crosslinking between molecular chains of the sample without the viscosity stabilizer was degraded. This led to changes in the structure and solvent easily penetrated into the NR structure.

Crosslinking density (ν) of vulcanized NR samples; a) WNR and b) PNR. S means sulphur vulcanization and STA means viscosity stabilizer
For PNR with a viscosity stabilizer, the ν value was constant until a curing time of 32 min (2 × t90), while PNR without a viscosity stabilizer began to deteriorate (Fig. 9b) and the ν value decreased from the curing time of 16 (t90) min until 48 min (3 × t90). From the result of crosslinking density obtained from crosslinked WNR and PNR, we found that the viscosity stabilizer not only affected the network formation of the non-rubber components but also can prevent reversion in NR vulcanizate.
The molecular weight between crosslinks (Mc) indicated the occurrence of crosslinkages between NR chains. If the NR sample had more crosslinks, a low Mc was observed. In other words, low Mc values mean high crosslinking density in the sample. The Mc results are shown in Fig. 9. WNR gave a lower Mc value than PNR because PNR had a very low content of proteins and lipids which act as the accelerator in the vulcanization reaction and can cause gel formation in NR. In this case, the PNR samples have a lower crosslinked network or crosslinking density than WNR. The crosslink between the molecular chains of PNR were lower than those of WNR, leading to its higher Mc.
Considering the effect of a viscosity stabilizer, we found that WNR samples with and without a viscosity stabilizer had similar Mc values at curing times of 4 and 8 min. However, the Mc of WNR without a viscosity stabilizer was slightly higher at 12 min. WNR with a viscosity stabilizer at 12 min had a higher ν value than without, indicating that its Mc value was lower. In the case of PNR, the result of Mc had the same tendency as the crosslinking density result (Fig. 9b). Mc values of PNR with a viscosity stabilizer remained the same at a curing time of 16 and 32 min and then increased at 48 min because the crosslinking density at 48 min was the lowest. For PNR without a viscosity stabilizer, the Mc values increased as the curing time increased. This result suggested that the viscosity stabilizer affected the gel formation of non-rubber components in NR and the occurrences of crosslinking between molecular chains of NR were stabilized by the viscosity stabilizer.
Regarding the mechanical properties, Table 4 shows the results from the tensile test of vulcanized WNR and PNR. Their modulus was measured at 100% and 300% strain. The strength of NR depends on the crosslinking density. WNR showed higher modulus at 100% and 300% strain than PNR because WNR had more crosslinking density than PNR. Since WNR was stronger and tougher, therefore, WNR needed more force during stretching than PNR. This result is in good agreement with the previous results [21,22,23].
The results of the modulus at 100% of the crosslinked NR samples with and without a viscosity stabilizer are shown in Table 4. The modulus result of WNR and PNR either with or without a viscosity stabilizer was related to their crosslinking density. At 100% strain, the same modulus of WNR with and without a viscosity stabilizer for all curing times was observed in Table 4. In the case of PNR at 100% strain, the modulus at a curing time of t90 and 2 × t90 with and without a viscosity stabilizer was the same, but at 3 × t90, PNR with a viscosity stabilizer had a higher modulus than the counterpart without a viscosity stabilizer. This result is related to the crosslinking density result where PNR with a viscosity stabilizer at a curing time 3 × t90 had more crosslinking density and a higher modulus than those without. At 300% strain, the same tendency as 100% strain was observed. The results supported that a viscosity stabilizer affects non-rubber components in the formation of a gel network and it can also prevent the reversion in NR.
Conclusions
Proteins and lipids had a remarkable effect on storage hardening of NR. The dynamic gel networks from the reaction of proteins and lipids with the α- and ω-terminals of rubber chains increased after prolonged storage. The viscosity stabilizer restrained the intermolecular crosslinks between non-rubber components of the rubber chains, resulting in an almost constant Mooney viscosity and molecular weight. For the crosslinked NR samples, the viscosity stabilizer prevented the reversion. The mechanical properties also support the fact that WNR had a higher crosslinking density and network structure than PNR, leading to a greater observed modulus. The crosslinked NR samples were stabilized by the viscosity stabilizer.
References
Amnuaypornsri Y, Sakdapipanich J, Toki S, Hsiao B, Ichikawa N, Tanaka Y (2007) Paper#16, the meeting of the fall rubber division, ACS, Cleveland, Ohio, Oct 16–18, 2007, 81, 753–765
Smitthipong W, Suethao S, Shah D, Vollrath F (2016) Interesting green elastomeric composites: silk textile reinforced natural rubber. Polym Testing 55:7–24
Li L, Smitthipong W, Zeng H (2015) Mussel-inspired hydrogels for biomedical and environmental applications. Polym Chem 6:353–358
Chollakup R, Smitthipong W, Chworos A (2014) DNA-functionalized polystyrene particles and their controlled self-assembly. RSC Adv 4:0648–30653
Chollakup R, Smitthipong W (2012) Chemical structure, thermal and mechanical properties of poly (nucleotide)–cationic amphiphile films. Polym Chem 3:2350–2354
Prasertsri S, Rattanasom N (2012) Fumed and precipitated silica reinforced natural rubber composites prepared from latex system: mechanical and dynamic properties. Polym Testing 31:593–605
Wang S, Zhang X, Wua X, Lu C (2016) Tailoring percolating conductive networks of natural rubber composites for flexible strain sensors via a cellulose nanocrystal templated assembly. Soft Matter 12:845–852
Flory PJ, Rehner JJ (1943) Statistical mechanics of cross-linked polymer networks I. rubberlike elasticity. J Chem Phys 11:512–520
Vaysse L, Beuve JS, Bonfils F (2012) SEC-MALS study of dynamic structuring of natural rubber: comparative study of two Hevea brasiliensis genotypes. J Appl Polym Sci 124:570–1577
Yunyongwattanakorn J, Tanaka Y, Kawahara S, Klinklai W, Sakdapipanich J (2003) Effect of non-rubber components on storage hardening. Rubber Chem Technol 76:1228–1240
Nakaramontri Y, Pichaiyut S, Wisunthorn S, Nakason C (2017) Hybrid carbon nanotubes and conductive carbon black in natural rubber composites to enhance electrical conductivity by reducing gaps separating carbon nanotube encapsulates. Eur Polym J 90:467–484
Smitthipong W, Nardin M, Schultz J, Nipithakul T, Suchiva K (2004) Study of tack properties of uncrosslinked natural rubber. J Adhesion Sci Technol 18:1449–1463
Ngolemasango F, Ehabe E, Aymard C, Sainte-Beuve J, Nkouonkam B, Bonfils F (2003) Role of short polyisoprene chains in storage hardening of natural rubber. Polym Int 52:1365–1369
Nimpaiboon A, Sriring M, Sakdapipanich JT (2016) Molecular structure and storage hardening of natural rubber: insight into the reactions between hydroxylamine and phospholipids linked to natural rubber molecule. J Appl Polym Sci 133:43753
Nawamawat K, Sakdapipanich JT, Ho CC (2010) Effect of Deproteinized Methods on the Proteins and Properties of Natural Rubber Latex during Storage. Macromol Symp 288:95–103
Kawahara S, Ruangdech J, Isono Y, Hikosaka M, Tanaka Y (2003) Effect of non-rubber components on the crystallization behavior or natural rubber. J Macromol Sci 42:761–771
Smitthipong W, Nardin M, Schultz J (2008) Effect of bulk and surface properties on adhesion of rubbers. Kasetsart J (Nat Sci) 42:318–324
Amnuaypornsri S, Nimpaiboon A, Sakdapipanich J (2009) Role of phospholipids and proteins on gel formation and physical properties of NR during accelerated storage. Kaut Gummi Kunsts 62:88–92
Tarachiwin L, Sakdapipanich J, Ute K, Kitayama T, Tanaka Y (2005) Structural characterization of alpha-terminal group of natural rubber. 1. Decomposition of branch-points by lipase and phosphatase treatments. Biomacro 6:1858–1863
Toki S, Sics I, Ran S, Liu L, Hsiao BS, Murakami S, Tosaka M, Kohjiya S, Poomprsdub S, Ikeda Y, Tsou AH (2004) Strain-induced molecular orientation and crystallization in natural and synthetic rubbers under uniaxial deformation by in-situ synchrotron x-ray study. Rubber Chem Technol 77:317–335
Alig I, Pötschke P, Lellinger D, Skipa T, Pegel S, Kasaliwal GR, Villmow T (2012) Establishment, morphology and properties of carbon nanotube networks in polymer melts. Polym 53:4–28
Suchiva K, Kowitteerawut T, Srichantamit L (2000) Structure properties of purified natural rubber. J Appl Polym Sci 78:1495–1504
Xu L, Huang C, Luo M, Qu W, Liu H, Gu Z, Jing L, Huang G, Zheng J (2015) A rheological study on non-rubber component networks in natural rubber. RSC Adv 5:91742–91750
Smitthipong W, Nardin M, Schultz J, Suchiva K (2007) Adhesion and self-adhesion of rubbers, crosslinked by electron beam irradiation. Int J Adhes Adhes 27:352–357
Acknowledgements
This work was financially supported by Kasetsart University Research and Development Institute (KURDI), Kasetsart University, Thailand. The authors wish to thank Specialized center of Rubber and Polymer Materials in agriculture and industry (RPM), Faculty of Science, Kasetsart University for the publication support. For characterization of the rubber samples, the authors wish to thank the Centre for Rubber Research, Faculty of Science, Mahidol University (Thailand), Faculty of Science, Kasetsart University (Thailand) and Rubber Research Institute of Thailand.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chollakup, R., Suwanruji, P., Tantatherdtam, R. et al. New approach on structure-property relationships of stabilized natural rubbers. J Polym Res 26, 37 (2019). https://doi.org/10.1007/s10965-018-1685-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1685-1