
Abstract
In this study, poly(vinyl pyrrolidone) (PVP) polymeric fibers were obtained by the solution blow-spinning (SBS) technique using PVP of low molecular weight. The fabrication of nano-microfibers of polymers with low molecular weights by using the SBS or electrospinning (ES) techniques is not common. Most theoretical studies suggest that only high-molecular-weight polymers can be produced because of their rheological properties. The influence of solution parameters (concentration, solvent volatility, and viscosity) and processing parameters (injection rate, collector rotation, gas pressure, and needle size protuberance) on the formation, morphology, and physical properties of the PVP fibers was investigated using the images obtained by scanning electron microscopy (SEM), thermal analysis (DSC/TG), x-ray diffraction (XRD) patterns, and Fourier-transform infrared spectroscopy (FTIR) spectra. The results showed no significate changes in the polymer properties because of fiber processing. Additionally, the fiber diameter frequency distribution was analyzed for each condition studied, and the behaviors of the fiber diameters with higher occurrences were evaluated as a function of the variables in the study, revealing that there is no simple relationship between the fiber diameter and processing conditions.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
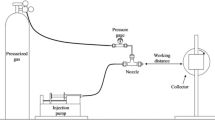
Solution blow-spinning (SBS) technique [1, 2] has been successfully employed as an alternative to the well-established electrospinning technique for producing micro- and nanofibers [3, 4]. The main advantages of SBS are its low cost, easy implementation, the deposition of fibers onto any type of substrate or collector, and a fiber production rate many times higher than that obtained by using techniques conventionally employed in the production of fibers. Usually, an injection rate of 1–6 μL/min is recorded for the electrospinning technique against that of 10–140 μL/min for SBS [3,4,5,6,7].
In order to successfully obtain blow-spun polymeric fibers, two classes of parameters must be observed: (i) solution parameters: viscosity, surface tension, concentration, molecular mass, and solvent volatility [8,9,10,11]; and (ii) processing parameters: solution injection rate, gas pressure, rotation speed of the collector, working distance, needle diameter, and insertion of the needle from the gas tip [12,13,14,15].
Among the materials produced into micro- and nanofibers, we can highlight poly(vinyl pyrrolidone) (PVP), (C6H9NO)n, a synthetic polymer whose molecular weight can range from 6000 to 3,000,000, with physicochemical properties depending on its molecular weight. Usually, only PVP polymers of high molecular weights, around 1,300,000 Mw, have been extensively used in the production of fibers via electrospinning associated with various other types of materials [16,17,18]. These polymers stand out for having good adhesion to substrates, good complexing properties, low chemical toxicity, high hygroscopicity, excellent physicochemical compatibility, biocompatibility, and good solubility in water and several organic solvents, characteristics that allow a wide range of applications, besides their low cost when compared to other polymers [11, 19]. Additionally, PVP is an excellent polymer matrix compound for regulating the rheological properties of precursor fiber solutions, thus enabling the pulling and formation of composite fibers, polymer blends, and biofunctional or ceramic materials [16,17,18,19].
Although there are many studies on PVP fiber formation, it always plays a secondary role in the fiber formation. Only a few studies are devoted to the manufacturing of pure PVP fibers [16,17,18,19,20], and these studies are mainly focused on the influence of the solvent and/or solution concentration on the morphology of the obtained fibers and use PVP with a high molecular weight (1.300.000 MW). To our knowledge, this is the first report on the production of blow-spun PVP fibers by using PVP with a low molecular weight (360.000 MW) in SBS, the main advantage of which is the low cost of the raw material, considerably reducing the cost of producing large-scale fibers. Moreover, a low-molecular-weight material could be an interesting material to use as a polymer matrix for a controlled delivery system, in which the diffusion process could be improved. In this work, we extensively study the influence of solutions and process parameters on the formation of PVP fibers and structural and thermal properties of the formed fibers.
Materials and methods
Solution and fiber preparation
Poly(vinyl pyrrolidone) (PVP), (C6H9NO)n, with an average molecular weight of 360.000 was supplied in powder form from Sigma-Aldrich Chemical Company. The PVP was dissolved by adding PVP powder slowly to the solvent under constant stirring at room temperature. The solvent employed was a mixture of isopropyl alcohol (IA) (C3H8O) and deionized water in different volume:volume percent (vol:vol%) proportions, and the polymer concentration was varied from 0.04 to 0.10 g/mL to evaluate the influence of the polymer concentration and solvent nature on the fiber formation.
The fiber formation tests were performed by varying many parameters, such as the polymer concentration solvent mixture AI/H2O (vol:vol), injection rate, gas pressure, collector rotation, needle (22 G) protuberation, and the work distance between the collector and the needle.
Sample characterization
The solutions’ viscosities were evaluated in a Brookfield rheometer, DV-II + Pro model, with concentric geometry, as a function of temperature from 0 to 60 °C. The crystalline properties were investigated by an XRD diffraction technique in a Shimadzu Corp., XRD-6000 model with Bragg-Brentamo geometry, CuKα radiation λ = 1.54056 Å, from 5 to 60° and in a continuous step mode with steps of 0.02°.
SEM (scanning electron microscopy) images were obtained in a Carl Zeiss, model EVO-LS15, 8–10 kV with an SE detector. Fiber diameters were measured by using IMAGE J software (National Institutes of Health, USA), by analyzing 100 random fibers from each image.
The thermal properties of the produced fibers were studied by DSC (differential scanning calorimetry) in a TA instruments, model MDSC 2920. The measurement conditions employed were a nitrogen atmosphere flux of 50 cm3/min, a temperature interval of −70 to 300 °C with heating rates of 2.5, 5.0, 10.0, and 20 °C/min. Additionally, thermogravimetric analysis (TGA) was performed in a TA instruments, SDT Q600 model, in an oxygen atmosphere flux of 100 mL/min, with a temperature interval of 30−1000 °C and a heating rate of 10.0 °C/min.
Results and discussion
The authors recommend that the supplementary material must be analyzed to better understanding of the results discussed below. The study of the solution’s rheological properties is one of the initial characterizations that must be done to evaluate the solution’s potential for fiber processing. Understanding the solution’s behavior is important to know the continuous flow of the fluid, under an external shear stress, in addition to the degree of polymer chains folding in the solution [21]. The rheological characterization of PVP solutions as a function of concentration (see supplementary material) and temperature showed the typical behavior of Newtonian fluids.
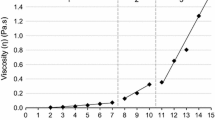
The viscosity values for the solutions investigated in this study vary from a maximum value of 0.73 Pa·s at a 0.12 g/mL concentration to a minimum of 0.05 Pa·s at a 0.04 g/mL concentration. In general, the viscosity (η) and concentration (C) of the polymer solution are related by expression to the fourth power. This dependence is valid for a wide concentration range, which can be divided into diluted, semi-diluted and concentrated regimes [21]. For the same concentration regime, the behavior of η vs C is approximately linear [22]. The linear behavior observed for the viscosity. The linear behavior observed for the viscosity solutions as a function of concentration, Fig. 1a, indicates that the solutions are within the same concentration regime, i.e., semi-dilute regime [9, 21, 22]. The dependence of the solution’s viscosity on the solvent nature is shown in Fig. 1b; a vertiginous decay was observed by increasing the amount of IA in the solution, and this behavior was followed by a small increase of the solution’s viscosity, which was observed for pure isopropyl alcohol.

PVP solution Viscosity and Fiber diameter behavior as function of (a) concentration, and (b) Solvent mixtures
Influence of solution concentration on fiber formation
The solution concentration and the solvent volatility can be considered the main parameters for successfully producing SBS fibers. Initially, we studied PVP solutions produced by dissolving the polymer in IA. The influence of the solution’s concentration was evaluated in the range of 0.04–0.12 g/mL. For the minimum concentration (0.04 g/mL), a film with a small amount of fibers was observed. By increasing the concentration, the production of fibers was improved, and their morphology became better defined.
A detailed analysis of the fibers deposited revealed that PVP dried particles were also deposited during the SBS processing, which can be attributed to the high volatility of the solvent. It contributed to forming the dried particles at the needle tip, and they were then ejected, reaching the fiber mats in the collector. The fiber diameter analysis revealed a micrometer formation of the fibers and no direct dependence between the fiber diameter (considering the higher occurrence of fiber diameters on the histogram) and the solution concentration, as shown in Fig. 1a. The fiber diameter distribution function (histogram) showed a monomodal behavior with well-defined maxima and variable widths for the conditions analyzed.
This results point to the existence of a processing window in terms of the concentration of the solution, limited by the range of 0.04–0.12 g/mL, which was identified. Outside of this range, it was not possible to obtain PVP fibers; below 0.04 g/mL, only a continuous film could be deposited, and above 0.12 g/mL, the gas pressure was not strong enough to pull fibers, or even to form the Taylor cone.
Influence of solvent on fiber formation
The influence of the mixture of deionized water and isopropyl alcohol, which mainly affects the solvent volatility, on the production of fibrous mats was investigated. The concentration of the PVP employed was 0.10 g/mL, and this choice was based on the results obtained up to that point, as it exhibits smooth and homogeneous fibers. The proportions studied were based on the v:v% mixture of IA and deionized water, from 50:50 (IA/H2O) to pure IA (100:0).
The changes in viscosity of the solution, shown in Fig. 1b, are directly associated with the degree of chain folding in the solution [22]. The results obtained point to a higher degree of chain folding when the amount of water present in the system increased. This observation, associated with the requirements for fiber pulling, indicated that the 50:50 AI/H2O condition would present greater difficulty in the jet stretching and the consequent fiber formation, associated with the significant decrease in volatility of the solution, made the fiber drying and morphology preservation after the fiber stretching more difficult.
The results obtained for solvent mixtures revealed that there was a maximum water limit for fiber formation to occur. For the volume ratio of 50:50 (IA/H2O), it was not possible to obtain fibers deposited on the collector. The low water volatility produced only a continuous, thick, and still moist film. Few fibers were formed for the 60:40 (IA/H2O) mixture. The fibers formed were sparse and in small quantities. The presence of defects in the mats was troublesome, and just below the fibers it was possible to notice the formation of a continuous film, due mainly to the large presence of water in the system that caused the fibers to melt.
A considerable increase in the number of fibers formed was observed for the ratio of 70:30 (IA/H2O), as shown in Fig. 2a. The fibers formed were cylindrical, with a smooth and homogeneous appearance, free of dry material precipitated on the surface. Some joints in certain points of contact were demonstrated, associated with the amount of water in the fibers causing the fibers to melt. The fiber diameter distribution function, Fig. 2b, showed a maximum distribution centered around 0.8 μm, presenting a wide and monomodal distribution.

SEM image (a) of PVP fiber (0.1 g/mL) produced by using a mixture of ratio of 70:30 (IA/H2O) and (b) respective histogram
Similar results was observed in other mixtures of (IA/H2O) analyzed; 80:20, 90:10, and pure IA. The main difference observed can be associated with the solvent properties. A more detailed analysis of the fiber morphology revealed that for ratios above 80:20, a dried material started to be deposited on the fiber mats. The fiber diameter measured increased exponentially as the water concentration decreased.
Solution injection rate influence on fiber formation
The solution injection rates were varied from 0.06 to 0.15 mL/min. A dense fiber mat, presenting fibers with a cylindrical, smooth, and homogeneous morphology, could be obtained for all conditions analyzed. Only a few defects associated with the dried PVP particles, or even entire drops expelled from the needle tip, were identified in all samples produced. The fiber diameter frequency distribution was a monomodal type, and became narrower as the injection rate increased. The values of fiber diameters varied from 0.9 to 1.3 μm. There was no clear mathematical relationship between the fiber diameter and the solution injection rate.
Gas pressure influence on fiber formation
Subtle variations in the morphology of the fiber mats were obtained by changing the gas pressure; the fiber density seemed to increase with the increasing the gas pressure. The gas flow also seemed to influence the fiber diameter distribution, the maximum fiber diameter values varied from 700 to 900 nm. The fiber diameter function proved to be broad and well defined for the studied conditions, presenting a monomodal characteristic. The smallest diameter observed was 750 nm, obtained by using a gas pressure of 160 kPa. In general, the same morphology observed before was obtained by changing the gas pressures; cylindrical, smooth, and continuous fibers, with the presence of few melting points and dry particles deposited on them. Once again, no direct relationship between the fiber diameter and the gas pressure could be demonstrated.
Needle size protuberation influence on fiber formation
The needle protuberation refers to the insertion distance of the needle tip in the air flow region, measured from the base of the nozzle. The region in which the needle tip is inserted acts on two distinct and simultaneous parameters relating to the fiber pull, jet stability and drag force. Some defects in the fibrous mat were demonstrated for lengths less than 5.0 mm. The diameter distribution function was broad with monomodal characteristic, the fiber diameter presented values ranging from 600 nm to 1.0 μm. The lowest dispersion of the fiber diameter was observed at 5.0 mm.
Collector rotation influence on fiber formation
The results obtained by varying the collector rotation speed from 150 to 350 rpm showed no influence on the fiber orientation or morphology. The random fibers were deposited on the collector, with fiber diameter showing a small variation around 1.0 μm. The fiber diameter showed a parabolic dependence on the collector rotation speed. The fiber diameter values increased from 900 nm at 150 rpm up to 1.1 μm at 250 rpm, then decreased to 900 nm at 350 rpm.
Characteristic defects observed on PVP fibers obtained by SBS
Fig. 3 shows the most common defects observed in the PVP fibers obtained. It was possible to observe the occurrence of four types of defects, which were present in different experimental conditions studied, not being characteristics of a certain parameter, and therefore related to the nature of some mechanism involved in the process.

Different morphological “defects” presented on PVP fibers (a) beads, (b) dried particles, (c) entire drops and (d) melted fibers
The smallest observed occurrence was the presence of beads in the formation of fibers as shown in Fig. 3a, which were observable in only two conditions, and in a small amount. This was interesting because numerous variables were analyzed and the beads formation was unaffected, which is in opposition with the results described in the literature, where the beads formation is very common in the electrospinning technique [10].
Another defect commonly found was the ejection of dry material (PVP), as shown in Fig. 3b. The ejection of PVP in the form of a dry particulate was common in most of the analyzed situations, the nature of the formation of this defect was mainly related to the solvent volatility, solution flow, and gas pressure.
Figure 3c exemplifies a typical “blown drop” type formation, where an entire drop is expelled from the needle tip and breaks on the fiber mat surface. This defect was quite common in the formation of polymer mats and its occurrence was mainly associated with high injection rates, low gas pressure, high water content, low viscosity, and low concentration of polymer in the solution. The nature of the formation was basically identified by the inability of the system to provide sufficient pulling of the fibers, or the stretching of the jet or polymer solution, resulting in a large amount of liquid remaining at the tip of the needle, which was eventually expelled as a drop. When colliding with the target, this drop exploded, forming the structure observed in Fig. 3c and involving numerous fibers, thus damaging the morphology of the polymeric mats as a whole.
The melting of fibers shown in Fig. 3d occurred mainly after their formation, i.e., they were deposited and collected on the collector, and when in contact with each other, due to the action of solvent that remained inside the fiber, they melted or coalesced with each other.
Study of PVP fibers physical properties
During the fiber processing, the stretching suffered by the polymer chain associated with a high solvent volatility rate during the fiber formation could promove a polymer chain’s orientation in a particular direction. This arrangement could affect the crystallinity of the final polymer and has been reported as a recurring phenomenon in the processing of some materials in the form of fibers by electrospinning [3]. The diffractogram of PVP fibers, Fig. 4a, shows two relatively intense peaks situated at 2θ = 11.3° and 21.5°. Under these peaks, there is a large halo (solid line below the experimental points) due to x-ray dispersion, usually assigned to an amorphous fraction in semicrystalline polymers. We applied three Gaussian-type functions to adjust the experimental results, and the result obtained gave a good approximation of the experimental data, as can be observed in Fig. 4a by the continuous line passing through the experimental points.

PVP fiber mats produced with 0.1 g/mL and solvent mixture of 70:30 (IA/H2O) (a) XRD difratograma showing the Lorentzian functions used to peaks analyzed, (b) FTIR spectra of Fiber and Film for comparation between the main vibrational bands
The XRD deconvolution analysis revealed two broad and sharp peaks. Although PVP is an amorphous polymer, for comparative purposes, and if there were any orientation changes on the polymer chains due to the inherent processing conditions, the alignment of the fibers could be studied by analyzing the changes in these two peaks. By analyzing this peaks of different PVP fibers obtained by using solvent mixtures of 90:10, 80:20, and 60:40, the relative intensity between the first peak and the second peak was substantially the same. The peak positions had small variations as a function of the amount of isopropyl alcohol present in the solvent. A large difference was observed for the 70:30 composition whose relative intensity peaks value was 1.96 times higher than that of the fiber obtained with isopropyl alcohol alone (1.04). The results point to no influence of the fiber processing and solvent volatility on the fibers’ crystallinity.
FTIR analysis of the PVP fibers, shown in Fig. 4b, was investigated by comparison with that of a PVP casting film. Both FTIR spectra showed no significant changes between the casting film and the SBS fiber. The main PVP bands were observed at around 1660 cm−1, ascribed to the stretching vibration of C=O in the carbonyl groups; the peak around 880 cm−1 could be assigned to the breathing vibration of the pyrrolidone ring, and finally, the presence of hydroxyl and C-N stretching modes was observed around 1264 cm−1 [18, 21].
Figure 5a shows the results of the TG and differential thermal analysis (DTA) analysis for the sample of PVP fibrous mat obtained with a concentration of 0.10 g/mL and an AI/H2O ratio of 100:0. The TG results suggest that the mass loss of the material began at approximately 280 °C and continued until approximately 600 °C, and this behavior is in accordance with those described in the literature for PVP. Considering the percentage of mass present at the plateau, located in the range of 100–280 °C, where the sample was thermally stable, after the sample underwent a thermal decomposition process, we recorded a mass loss of 92.20%. Remaining at the end was a residue of only 1.07% at temperatures above 600 °C.

Thermal characterization of PVP fiber mats produced with 0.1 g/mL and solvent mixture of 70:30 (IA/H2O) (a) TG/DTA analysis and (b) DSC analysis at different heating rates
The degradation curve of PVP presented a complex behavior, with at least four distinct thermal events occurring during the heating process of the polymer. The first event, with a small loss of mass of 6.73%, occurred at the beginning of the heating process and ceased at a temperature of around 100 °C, possibly being associated with the loss of water or volatile products, remembering that PVP is a highly hygroscopic material. Corresponding to this event we observed, in the DTA graph, a broad and low intensity endothermic peak in this temperature range.
A second thermal event started at about 260 °C with a mass loss of approximately 16.43%, which ceased around 397 °C. This event was demonstrated in the DTA graph by a relatively intense narrow exothermic peak centered at 360 °C. Deconvolving the peaks of the DTA analysis with Lorentzian functions, as shown in the upper inner corner of Fig. 5a, we can calculate an approximate area for this peak, thus obtaining an estimated ΔH value at 273.83 J/g.
The third and most pronounced thermal event was marked by a mass loss of approximately 46.61% in the range of 397–457 °C. The pronounced peak of the DTA analysis appears to be a result of more than one exothermic process. This narrower, more intense peak was centered at 419 °C; the area below the peak, calculated as previously described, gave an estimated ΔH value of 945.85 J/g. The fourth process, demonstrated in the interval from 457 °C to 600 °C, presents a mass loss of 29.03%. The peak associated with this process shows low intensity and wide width, being centered around 505 °C, and the area below the peak was calculated at an estimated ΔH value of 1418.0 J/g.
In part, our results are consistent with the results observed in the literature for pure PVP. This process of degradation of PVP was attributed only to two thermal events, the first one from 250 °C to 440 °C with a loss of mass of 70%, and the second from 440 to 550 °C, with a mass loss of 30% attributed to thermo-oxidative processes where the occurrence of active degradative species was promoted by the presence of oxygen in the atmosphere.
Figure 5b illustrates the results obtained from differential scanning calorimetry (DSC) analyses for pure commercial PVP and fibrous mats produced using 0.10 g/mL PVP with different IA/H2O mixtures in a nitrogen atmosphere. All the analyzed samples presented similar behaviors in the studied range (−50 to 300 °C), where only a single endothermic peak can be identified in the range of 100–150 °C. This peak, possibly due to the hygroscopic nature of PVP, refers to the release of water, which is adsorbed into the polymer. Its shape and position have undergone a slight variation in function for the solvent, but the nature of the phenomenon remained the same.
The variation in the shape of the peak and in its area is associated to the enthalpy change required for the adsorption of water molecules, indicating that the amount of water present in each sample varied. However, we cannot determine if this phenomenon was inherent to the process, because water molecules can also be absorbed from the atmosphere by the material. Tests varying the heating rate (results not shown here) revealed that little or no change occurred in the peak or shape position, shown in Fig. 5b.
Conclusions
Sub-micrometer fibers of a low-molecular-weight PVP were produced by SBS after the proper choice of solution concentration and solvent mixture was made, showing a cylindrical, smooth, and continuous morphology. The usual defects observed in electro-spun fibers could also be identified on the SBS fibers. Concerning the solution parameters, the solvent nature and solution concentration were revealed to be the most important and decisive parameters in blow-spun fiber production. The SBS fiber processing window proved to be barely influenced by changes in the processing parameters. Once the processing window was achieved, no significant changes on fiber diameter or fiber formation could be observed. In fact, our results point to no direct mathematical relationship between the fiber diameter and the processing parameters.
References
Medeiros ES, Glenn GM, Klamcznyski AP, Orts WJ, Mattoso LHC (2009) Solution blow spinning: A new method to produce micro- and nanofibers from polymer solutions. J Appl Polym Sci 113:2322–2330
Cena CR, Larios GS, Bica MRR, Canassa TA, Freitas GQ, Torsoni GB (2015). Rev Bras de Fís Tecn Apl 2:32
Li D, Xia Y (2004) Electrospinning of Nanofibers: Reinventing the Wheel? Adv Mater 16:1151–1170
Cena CR, Behera AK, Behera B (2016) Structural, dielectric, and electrical properties of lithium niobate microfibers. B J of Adv Ceram 5:84–92
Cena CR, Torsoni GB, Zadorosny L, Malmonge LF, Carvalho CL, Malmonge JA (2017) BSCCO superconductor micro/nanofibers produced by solution blow-spinning technique. Ceram Inter 43:7663–7667
Guerrini LM, Branciforti MC, Bretas RES, Oliveira MP (2006) Eletrofiação do poli (álcool vinílico) via solução aquosa. Polím. Ciên. e Tecn. 16:286–293
Silva TH, Oliveira JE, Medeiros ES (2015) Obtenção de micro e nanofibras de PVC pela técnica de Fiação por Sopro em Solução. Polím Ciên e Tecn 25:229–235
McKee MG, Wilkes GL, Colby RH, Long TE (2004) Correlations of Solution Rheology with Electrospun Fiber Formation of Linear and Branched Polyesters. Macromolecules 37:1760–1767
Oliveira JE, Moraes EA, Costa RGF, Afonso AS, Mattoso LHC, Orts WJ, Medeiros ES (2011) Nano and submicrometric fibers of poly(D,L-lactide) obtained by solution blow spinning: Process and solution variables. J. of Appl. Polym. Sci. 122:3396–3405
Fong H, Chun I, Reneker DH (1999) Beaded nanofibers formed during electrospinning. Polym 40:4585–4592
Chuangchote S, Sagawa T, Yoshikawa S (2009) Electrospinning of poly(vinyl pyrrolidone): Effects of solvents on electrospinnability for the fabrication of poly(p-phenylene vinylene) and TiO2nanofibers. J of Appl Polym Sci 114:2777–2791
Bhardwaj N, Kundu SC (2010) Electrospinning: A fascinating fiber fabrication technique. Biotechn Adv 28:325–347
Costa RGF, Oliveira JE, Paula GF, Picciani PHS, Medeiros ES, Ribeiro C, Mattoso LHC (2012). Polím. Ciên. e Tecn. 22:12
Feng JJ (2002) The stretching of an electrified non-Newtonian jet: A model for electrospinning. Phys Fluids 14:3912–3926
Cui W, Li X, Zhou S (2007) Investigation on process parameters of electrospinning system through orthogonal experimental design. J Weng J Appl Polym Sci 103:3105–3112
Yang Q, Li Z, Hong Y, Zhao Y, Qiu S, Wang CE, Wei Y (2004) Influence of solvents on the formation of ultrathin uniform poly(vinyl pyrrolidone) nanofibers with electrospinning. J Polym Sci 42:3721–3726
Mikheev AY, Kanev IL, Morozova TY, Morozov VN (2013) Water-soluble filters from ultra-thin polyvinylpirrolidone nanofibers. J Memb Sci 448:151–159
Bonan RF, Bonan PRF, Batista AUD, Perez DEC, Castellano LRC, Oliveira JE, Medeiros ES (2017). J. Appl. Polym. Sci. 134:1
Buera MP, Levi G, Karel M (1992) Glass transition in poly(vinylpyrrolidone): effect of molecular weight and diluents. Biotech Prog 8:144–148
Kim K, Kim J, Shim H (2017) Fiber formation model for PVP (polyvinyl pyrrolidone) electrospinning. I. Critical voltage. Fiber Polym 18:493–501
Song YJ, Wang M, Zhang XY, Wu JY, Zhang T (2014). Nano Res Lett 9:1
Ji HM, Lee HW, Karim MR, Cheong IW, Bae EA, Kim TH, Islam MS, Ji BC, Yeum JH (2009) Electrospinning and characterization of medium-molecular-weight poly(vinyl alcohol)/highmolecular-weight poly(vinyl alcohol)/montmorillonite nanofibers. Colloid Polym Sci 287:751–758
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 9172 kb)
Rights and permissions
About this article

Cite this article
Cena, C.R., Silva, M.J., Malmonge, L.F. et al. Poly(vinyl pyrrolidone) sub-microfibers produced by solution blow spinning. J Polym Res 25, 238 (2018). https://doi.org/10.1007/s10965-018-1633-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1633-0