
Abstract
Two series of new triptycene-containing poly(ester-amide)s and poly(ester-imide)s were prepared from 1,4-bis(3-aminobenzoyloxy)triptycene with various aromatic dicarboxylic acids and dianhydrides, respectively. The synthesis of the poly(ester-amide)s was achieved by the phosphorylation polyamidation reaction by means of triphenyl phosphite and pyridine, and the synthesis of the poly(ester-imide)s included ring-opening polyaddition to give poly(amic acid)s followed by thermal imidization and chemical imidization to polyimides. All the poly(ester-amide)s and most of the poly(ester-imide)s presented good solubility in many organic solvents and could be solution-cast into transparent and flexible films. These poly(ester-amide)s and poly(ester-imide)s displayed discernible glass-transition temperatures (Tgs) between 242 and 298 °C in the DSC traces and showed moderate thermal stability with 10 wt% loss temperatures above 464 °C in nitrogen or air. These highly optically transparent polymer films possess an ultraviolet-visible absorption cut-off wavelength (λ0) down to 344 nm. 1,4-Bis(4-aminobenzoyloxy)triptycene was also synthesized and used for polymer synthesis; however, less favorable results were obtained.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Aromatic polyamides and polyimides are well known as high-performance polymers because of their outstanding thermal stability, high mechanical strength and modulus, and superior chemical resistance [1,2,3,4,5,6]. Noted examples include Kapton polyimide film and Nomex and Kevlar aramid fibers developed by Dupont in 1960s. They have a variety of applications in many different areas, including automotive, aerospace, defense, electronics, medical, and sport and safety equipment. However, most of the conventional aromatic polyamides and polyimides suffer processing difficulty due to their high melting temperatures (Tm) or glass-transition temperature (Tg) and limited solubility in most organic solvents, which are caused by their rigid backbones and strong interchain interactions. To improve their solution and melting processability, various attempts have been made via structural modifications [7,8,9]. One of the common approaches is the introduction of flexible linkages and/or packing-disruptive bulky groups in the polyamide and polyimide backbones [10,11,12,13,14,15]. In addition, most of aromatic polyimides usually show considerable coloration from pale yellow to dark brown, which hinders their use in flexible substrate for display device. In general, aromatic polyimides are thought to absorb in the visible region through formation of a charge-transfer complex (CTC) between the electron-donating diamine component and the electron-accepting dianhydride component [16, 17]. A common strategy on obtaining light-colored or colorless polyimide films is to use of dianhydrides with lower electron-accepting ability and diamines with lower electron-donating ability that can mitigate intra- and intermolecular CTC effect [18].
Triptycene is a rigid, bulky, three-dimensional molecular unit consisting of three benzene blades protruding from a single hinge [19, 20]. One of the structural features of triptycene is the intrinsic space between the benzene blades, known as the internal free volume. The use of triptycene moiety as a rigid and shape-persistent component is a method to introduce molecular scale free volume into a polymer film. Polymers with triptycene moiety might be interesting low-k dielectric materials owing to the high degree of internal free volume [21]. Additionally, when incorporated into polyimide backbones, the triptycene unit may hinder chain packing, resulting in tunable high fractional free volume for gas separation [22,23,24]. It has also been demonstrated that incorporation of both ether and triptycene units into the polymers backbones may enhance the solubility and processability of aromatic polyamides and polyimides without much loss in thermal stability [25,26,27,28,29]. Because of the unique structural feature of triptycene, synthesizing and charactering new polymers bearing triptycene moieties in the backbone are interesting enough to be investigated further. This work deals with the synthesis and characterization of two new triptycene bis(ester amine)s, namely 1,4-bis(3-aminobenzoyloxy)triptycene and 1,4-bis(4-aminobenzoyloxy)triptycene, and their derived poly(ester-amide)s and poly(ester-imide)s. It was expected that incorporation of three-dimensional triptycene moiety together with the ester linkage into the polymer backbone could improve the solubility, decrease the color intensity, and preserve moderate thermal stability.
Experimental
Materials
1,4-Dihydroxytriptycene (1) (mp = 340–342 °C) was synthesized according to a two-step synthetic route reported in the literature [25]. Anthracene (Alfa Aesar), p-benzoquinone (Acros), 3-nitrobenzoyl chloride (Acros), 4-nitrobenzoyl chloride (TCI), triethylamine (NEt3, Acros), 10% palladium on charcoal (Pd/C, Acros), triphenyl phosphite (TPP, TCI) and acetic anhydride (Acros) were used as received from the supplier. N,N-Dimethylacetamide (DMAc, Tedia), N,N-dimethylformamide (DMF, Tedia), pyridine (Py, Wako) and N-methyl-2-pyrrolidone (NMP, Tedia) were dried over calcium hydride for 24 h, distilled under reduced pressure, and stored over 4 Å molecular sieves in sealed bottles. Commercially obtained calcium chloride (CaCl2) was dried under vacuum at 180 °C for 3 h prior to use. The aromatic dicarboxylic acid monomers including terephthalic acid (4a, Wako), 4,4′-biphenyldicarboxylic acid (4b, Aldrich), 4,4′-dicarboxydiphenyl ether (4c, TCI), bis(4-carboxyphenyl) sulfone (4d, TCI) and 2,2-bis(4-carboxyphenyl)hexafluoropropane (4e, TCI) were used as received from commercial sources. The aromatic tetracarboxylic dianhydrides including pyromellitic dianhydride (PMDA; 6a, TCI), 3,3′,4,4′-biphenyltetracarboxylic dianhydride (BPDA; 6b, Oxychem), 4,4′-oxydiphthalic dianhydride (OPDA; 6c, Oxychem) and 2,2-bis(3,4-dicarboxyphenyl)hexafluoropropane dianhydride (6FDA; 6d, Hoechst Celanese) were purified by dehydration at 250 °C in vacuo for 3 h. Other reagents and solvents were used as received from commercial sources.
Monomer synthesis
1,4-Bis(3-nitrobenzoyloxy)triptycene (2) and 1,4-bis(4-nitrobenzoyloxy)triptycene (2′)
In a 250 mL three-neck round-bottom flask equipped with a stirring bar and a nitrogen gas inlet tube, 5.0 g (0.017 mol) of 1,4-dihydroxytriptycene (1) and 5.1 mL (0.037 mol) of triethylamine were dissolves in 100 mL of dried DMAc. A solution of 6.8 g (0.037 mol) of 3-nitrobenzoyl chloride in 20 mL DMAc was then added dropwise over a period of about 1 h. After complete addition, the reaction mixture was stirred at 60 °C for 12 h. The reaction mixture was then poured into 600 mL of water, and the precipitated product was collected by filtration, washed thoroughly by water and methanol, and dried in vacuum at 80 °C to give 8.35 g of the diester-dinitro compound 2 as a white powder in 84% yield.
IR (KBr): 1749 cm−1 (C=O stretch), 1522, 1348 cm−1 (−NO2 stretch), 1215, 1053 cm−1 (C − O stretch).

Similarly, diester-dinitro compound 2′ was synthesized from the condensation of 1,4-dihydroxytriptycene and 4-nitrobenzoyl chloride as white powder in 74% yield. IR (KBr): 1746 cm−1 (C=O stretch), 1523, 1345 cm−1 (−NO2 stretch), 1208, 1053 cm−1 (C − O stretch).

1,4-Bis(3-aminobenzoyloxy)triptycene (3) and 1,4-bis(4-aminobenzoyloxy)triptycene (3′)
In a 100 mL three-neck round-bottom flask equipped with a stirring bar, 1.3 g (0.002 mol) of diester-dinitro compound 2 and 0.15 g of 10% Pd/C were dispersed in 40 mL of DMF. The suspension solution was stirred at room temperature under a hydrogen atmosphere until the theoretical amount of hydrogen was consumed. The time taken to reach this stage was about 3 days. The solution was then filtered to remove Pd/C, and the obtained filtrate was poured into 250 mL of stirring water to give a white precipitate. The product was collected by filtration and dried in vacuum at 80 °C to give 0.77 g of bis(ester-amine) 3 as a white powder in 66% yield.
IR (KBr): 3467, 3372 cm−1 (amine −NH2 stretch), 1721 cm−1 (ester C=O stretch), 1204, 1085 cm−1 (ester C − O stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 5.56 (s, 4H, −NH2), 5.62 (s, 2H, Hb), 6.97 (dd, J = 7.7, 2.0 Hz, 2H, Hf), 7.00 (s, 2H, Ha), 7.02 (dd, J = 5.4, 3.1 Hz, 4H, Hc), 7.32 (t, J = 7.7 Hz, 2H, Hg), 7.40–7.50 (m, 6H, Hd + Hh), 7.51 (t, J = 2.0 Hz, 2H, He). 13C NMR (150 MHz, DMSO-d6, δ, ppm): 47.6 (C4), 114.7 (C13), 117.2 (C9), 119.2 (C11), 120.0 (C1), 124.1 (C7), 125.3 (C6), 129.1 (C3), 129.5 (C10), 139.1 (C8), 143.1 (C12), 144.1 (C5), 149.3 (C2), 165.1 (C=O). ESI-MS: 525.18 (M + H)+; Calculated exact mass for C34H24N2O4 = 524.17.

By a similar procedure, bis(ester-amine) 3′ was synthesized from hydrogen Pd/C-catalyzed reduction of diester-dinitro compound 2′ as a gray powder in 66% yield. IR (KBr): 3464, 3366 cm−1 (amine −NH2 stretch), 1720 cm−1 (ester C=O stretch), 1200, 1058 cm−1 (ester C − O stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 5.57 (s, 2H, Hb), 6.24 (s, 4H, −NH2), 6.75 (d, J = 8.7 Hz, 4H, Hf), 6.90 (s, 2H, Ha), 7.00 (dd, J = 5.4, 3.1 Hz, 4H, Hc), 7.38 (dd, J = 5.4, 3.1 Hz, 4H, Hd), 7.96, (d, J = 8.7 Hz, 4H, He). ESI-MS: 525.18 (M + H)+; Calculated exact mass for C34H24N2O4 = 524.17.

Polymer synthesis
Synthesis of poly(ester-amide)s
The synthesis of poly(ester-amide) 5a was used as an example to illustrate the general synthetic procedure used to produce the polyamides. A mixture of 0.314 g (0.60 mmol) of bis(ester amine) 3, 0.099 g (0.60 mmol) of terephthalic acid (4a), 0.15 g of anhydrous calcium chloride, 0.7 mL of triphenyl phosphite (TPP), 0.15 mL of pyridine, and 0.6 mL of NMP was heated with stirring at 120 °C for 3 h. The polymerization proceeded homogeneously throughout the reaction and afforded clear, highly viscous polymer solution. The resulting viscous solution was poured slowly with stirring into 160 mL of methanol, giving rise to a long-fiber precipitate. The precipitated product was collected by filtration, washed repeatedly with methanol and hot water, and dried to give a quantitative yield of poly(ester-amide) 5a. The inherent viscosity of polymer was 0.51 dL/g, measured in DMAc (containing 5 wt% LiCl) at a concentration of 0.5 g/dL at 30 °C.
IR spectrum of 5a (film): 3316 cm−1 (amide N − H stretch), 1735 cm−1 (ester C=O stretch), 1664 cm−1 (amide C=O stretch), 1196, 1061 cm−1 (ester C − O stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 5.74 (2H, Hb), 7.04 (4H, Hc), 7.10 (2H, Ha), 7.46 (4H, Hd), 7.73 (2H, Hg), 8.09 (2H, Hh), 8.24 (4H, Hi), 8.34 (2H, Hf), 8.79 (2H, He), 10.80 (2H, amide protons).

Synthesis of poly(ester-imide)s
The poly(ester-imide)s were prepared from various tetracarboxylic dianhydrides (PMDA, BPDA, OPDA or 6FDA) with bis(ester amine)s 3 and 3′, respectively, by a conventional two-step method via thermal or chemical imidization reaction. The synthesis of poly(ester-imide) 7d is described as an example. Into a solution of bis(ester amine) 3 (0.5412 g, 1 mmol) in 9.5 mL anhydrous DMAc in a 50 mL round-bottom flask, 0.4588 g (1 mmol) of 6FDA (6d) was added in one portion. Thus, the solid content of the solution is approximately 10 wt%. The mixture was stirred at room temperature for about 18 h to yield a viscous poly(ester-amic acid) solution with an inherent viscosity of 0.71 dL/g, measured in DMAc at concentration of 0.50 g/dL at 30 °C. The poly(ester-amic acid) film was obtained by casting from the reaction polymer solution onto a glass Petri dish and drying at 90 °C overnight. The poly(ester-amid acid) in the form of solid film was converted to the poly(ester-imide) film by successive heating at 150 °C for 30 min, 200 °C for 30 min, and 250 °C for 1 h. For chemical imidization method, 2 mL of acetic anhydride and 1 mL of pyridine were added to the poly(ester-amic acid) solution obtained by a similar process as above, and the mixture was heated at 100 °C for 1 h to effect a complete imidization. The homogenous polymer solution was poured slowly into an excess of methanol giving rise to a white precipitate that was collected by filtration, washed thoroughly with hot water and methanol, and dried. The inherent viscosity of the resulting poly(ester-imide) 7d was 0.47 dL/g, measured in DMAc at a concentration of 0.5 g/dL at 30 °C. A polymer solution was made by the dissolution of about 0.4 g of the poly(ester-imide) sample in 4 mL of hot DMAc. The homogeneous solution was poured into a glass Petri dish, which was placed in a 90 °C oven overnight for the slow release of solvent, and then the film was stripped off from the glass substrate and further dried in vacuum at 160 °C for 6 h.
The IR spectrum of 7d (film) exhibited characteristic imide and ester absorption bands at 1797 cm−1 (asymmetrical imide C=O stretch) and 1734 cm−1 (symmetrical imide and ester C=O stretch), 1202, 1115 cm−1 (ester C − O stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 5.74 (s, 2H, Hb), 6.98 (br, 4H, Hc), 7.07 (s, 2H, Ha), 7.42 (m, 4H, Hd), 7.81 (s, 2H, Hk), 7.87 (t, J = 7.7 Hz, 2H, Hg), 7.92 (d, J = 7.7 Hz, 2H, Hh), 8.01 (d, J = 7.7 Hz, 2H, Hj), 8.25 (d, J = 8.1 Hz, 2H, Hi), 8.38 (d, J = 7.7 Hz, 2H, Hf), 8.41 (s, 2H, He).

Measurements
Infrared (IR) spectra were recorded on a Horiba FT-720 FT-IR spectrometer. 1H spectra were measured on a Bruker Avance III HD-600 MHz NMR spectrometer with tetramethylsilane (TMS) as an internal standard. Mass spectroscopy was conducted on a JEOL JMS-700 mass spectrometer. The inherent viscosities were determined with a Cannon-Fenske viscometer at 30 °C. Molecular weights of the synthesized polymers were determined by gel permeation chromatography (GPC) using a polystyrene standard at 50 °C in NMP. The GPC apparatus is composed of a Jasco model PU-2080 HPLC pump, a Jasco model RI-2031 RI detector, and three Shodex GPC columns connected in series. Thermogravimetric analysis (TGA) was performed with a Perkin-Elmer Pyris 1 TGA. Experiments were carried out on approximately 3–5 mg of fully dried polymer film samples heated in flowing nitrogen or air (flow rate = 20 cm3/min) at a heating rate of 20 °C/min. DSC analyses were performed on a Perkin-Elmer DSC 4000 at a scan rate of 20 °C/min in flowing nitrogen. Ultraviolet-visible (UV-vis) spectra of the polymer films were recorded on an Agilent 8453 UV-visible spectrometer. Colorimetric data of the polymer films were measured on an Admesy Brontes colorimeter.
Results and discussion
Monomer synthesis
The bis(ester amine)s 3 and 3′ were synthesized via the synthetic route shown in Scheme 1. According to a reported method [25], 1,4-dihydroxytriptycene (1) was obtained in good yield starting from the Diels-Alder reaction of p-benzoquinone and anthracene and subsequent rearrangement reaction using acetic acid/HBr as catalyst. The intermediate diester-dinitro compounds 2 and 2′ were prepared by condensation of 1 with 4-nitrobenzoyl chloride and 3-nitrobenzoyl chloride, respectively, in DMAc in the presence of triethylamine. Bis(ester amine)s 3 and 3′ were prepared by the hydrogen Pd/C-catalyzed reduction of the diester-dinitro compounds 2 and 2′, respectively. IR, 1H NMR and 13C NMR spectroscopic techniques were used to identify the structures of the intermediate diester-dinitro compounds and the target bis(ester amine) monomers.

Synthetic route to the bis(ester amine) monomers 3 and 3’
Fig. S1 (Supporting Information) illustrates FT-IR spectra of all the synthesized compounds. The IR spectrum of compound 1 gives rise to a broad, strong absorption at about 3270 cm−1 ascribed to the O − H stretching vibration. The diester-dinitro compounds 2 and 2′ show the characterisic absorption of nitro groups at around 1522 and 1348 cm−1 (−NO2 asymmetric and symmetric stretching). After reduction, the characteristic absorptions of the nitro group disappear and the amino group shows the typical −NH2 stretching absorption pair in the region of 3300 to 3350 cm−1 as shown in the IR spectra of bis(ester-amine)s 3 and 3′. Conversion of a nitro group to an amino group shifts the C=O frequency from about 1750 to 1720 cm−1. All the diester-dinitro compounds and bis(ester amine)s exhibit C − O stretching absorptions in the 1000–1300 cm−1 range.
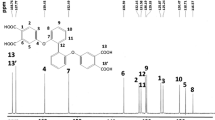
The 1H NMR, 13C NMR, and two-dimensional (2-D) NMR spectra of the bis(ester-amine) monomer 3 are compiled in Figs. 1 and 2. The resonance signal at 5.56 ppm ia ascribed to the aryl primary amino protons. The assignmernts of each proton and carbon are also given in the figures, and all the NMR spectroscopic data are consistent with the proposed mlecular structure of 3. Fig. S2 (Supporting Information) reproduces the 1H NMR and H − H COSY spectra of bis(ester-amine) 3′. The amino protons of 3′ resonate at a more downfield (δH = 6.24 ppm) than those of meta-isomer 3 because the electron-withdrawing ester carbonyl group is located at the para position to the amino group. Thus, the nucleophilicity (or basicity) for the amino groups of bis(ester-amine) 3 was expected to be stronger than that of its para-isomer 3′. In addition, mass analysis results of these two bis(ester-amine) monomers are consistent with the calculated exact mass.

(a) 1H NMR and (b) H-H COSY spectra of bis(ester-amine) 3 in DMSO-d6

(a) 13C NMR and (b) C-H HMQC spectra of bis(ester-amine) 3 in DMSO-d6
Synthesis of poly(ester-amide)s
According to the phosphorylation polyamidation technique described by Yamazaki and co-workers [30], two series of novel aromatic poly(ester-amide)s 5a − 5e and 5a’ − 5’e were synthesized from the polycondensation reactions of bis(ester amine) 3 and 3′, respectively, with five aromatic dicarboxylic acids 4a − 4e by using triphenyl phosphite (TPP) and pyridine as condensing agents (Scheme 2). The polymerization proceeded homogeneously throughout the reaction and afforded a clear, highly viscous polymer solution. All the 5 series polymers precipitated in a tough, fiber-like form when the resulting polymer solutions were slowly poured under stirring into methanol. However, the 5′ series polymers precipitated in a powder form. These two series of poly(ester-amide)s had inherent viscosities in the range of 0.51–0.57 dL/g for 5a − 5e and 0.29–0.44 dL/g for 5’a − 5’e, as shown in Table 1. The molecular weights of the synthesized poly(ester-amide)s were measured by GPC in NMP, and the relevant data are shown in Table 3. The number-average molecular weights (Mn) and weight-average weights (Mw) were recorded in the range of 20,600~28,100 and 31,900~46,100 for 5a − 5e, respectively, and 7900~14,300 and 10,000~24,100 for 5’a − 5’e, respectively, relative to polystyrene standards. All the meta-substituted poly(ester-amide)s can be solution-cast into flexible and strong films (as shown in inset of Scheme 2), indicating the formation of high molecular weight polymers. However, the para-substituted poly(ester-amide) films embrittled into pieces upon solution casting, possibly because of lower molecular weights. This result can be attributed to the lower nucleophilicity of the para-substituted bis(ester-amine) 3′ as mentioned above.

Synthesis of poly(ester-amide)s and the cast films of 5a − 5e from DMAc
The structures of the poly(ester-amide)s were confirmed by IR and NMR spectroscopy. Typical IR spectra for poly(ester-amide)s 5a and 5a’ are presented in Fig. S3 (Supporting Information). The characteristic absorption bands at around 3316 or 3367 cm−1 (amide N − H stretch) and 1664 or 1675 cm−1 (amide carbonyl stretch) confirm the formation of main chain amide linkages of 5a and 5a’. The 1H NMR and H-H COSY spectra of poly(ester-amide) 5a in DMSO-d6 are depicted in Fig. 3. The resonance signal at 10.56 ppm is ascribed to the amide protons. All the other resonance peaks agree well with the repeated unit structure of this poly(ester-amide).

(a) 1H NMR and (b) H-H COSY spectra of poly(ester-amide) 5a in DMSO-d6
Synthesis of poly(ester-imide)s
A series of triptycene-containing poly(ester-imide)s 7a − 7d were prepared from bis(ester-amine) 3 with various aromatic dianhydrides 6a − 6d by a conventional two-step procedure via the formation of poly(ester-amic acid)s, followed by thermal or chemical cyclodehydration (Scheme 3). As shown in Table 2, the poly(ester-amic acid) precursors had inherent viscosities in the range of 0.41–0.76 dL/g. The poly(ester-amic acid)s could be solution-cast into flexible thin films; however, all the poly(ester-amic acid) films embrittled during thermal cyclodehydration. Therefore, the poly(ester-amic acid) precursors were chemically dehydrated to the poly(ester-imide)s by treatment with acetic anhydride in the presence of pyridine. The poly(ester-imide)s 7a–C and 7b–C derived from PMDA and BPDA, respectively, precipitated during chemical imidization and were insoluble in available organic solvents, possibly because of the structural rigidity of the dianhydride components. In contrast, the poly(ester-imide)s 7c–C and 7d–C were soluble in the reaction medium and their solutions could be directly cast into transparent, flexible, and strong films, indicative of the formation of high-molecular-weight polymers. All the reactions of bis(ester-amine) 3′ with dianhydrides 6a − 6d produce low molecular weight poly(ester-amic acid)s which could not afford flexible and free-standing films. Therefore, no attempts were made to the preparation of poly(ester-imide)s from 3′. The poly(ester-imide)s soluble in NMP were characterized by GPC, and the relevant data are showed in Table 3. The number-average molecular weights (Mn) and weight-average weights (Mw) were recorded in the range of 12,000~21,100 and 17,400~37,200, respectively, relative to polystyrene standards. The poly(ester-imide)s generated by the thermal imidization method generally exhibited slightly lower inhererent viscosity and molecular weight than those obtained by the chemical imidization method. This implies that the ester linkages in the polymer chain may be hydrolyzed at elevated temperatures during thermal imidization.

Synthesis of poly(ester-imide)s 7a − 7d and their cast films from DMAc
Structural features of the poly(ester-imide)s were characterized by IR and NMR analysis. A typical set of IR spectral of poly(ester-imide) 7d and it poly(ester-amic acid) precursor are illustrated in Fig. S4. Poly(ester-imide) 7d exhibited characteristic imide group absorptions around 1795 and 1735 cm−1 (typical of imide carbonyl asymmetrical and symmetrical stretch), 1200 and 1115 cm−1 (ester C − O stretch). The ester carbonyl absorption was assumed to be buried in the strong imide carbonyl symmetrical stretch absorption at 1735 cm−1. 1H NMR and H-H COSY NMR spectra of poly(ester-imide) 7d in DMSO-d6 are presented in Fig. 4. All the aromatic protons resonate in the region of δ 5.5–8.5 ppm. Assignments of each proton are in good agreement with the structure of the repeating unit of 7d.

(a) 1H NMR and (b) H-H COSY spectra of poly(ester-imide) 7d in DMSO-d6
Polymer properties
The qualitative solubility properties of the polymers in several organic solvents at 10% (w/v) and the inherent viscosities are summarized at Table 1 and Table 2. All of the poly(ester-amide)s 5a − 5e presented good solubility; they were readily soluble in highly polar solvents such as NMP, DMAc, DMF, and DMSO at room temperature. Some of them were even soluble in less polar m-cresol and THF at room temperature or upon heating at 70 °C. In general, poly(ester-imide)s 7c and 7d showed a higher solubility than poly(ester-imide)s 7a and 7b because the former ones have a linking group between the phthalimide units. In particular, 7d exhibited a very good solubility in several organic solvents because of the presence of bulky hexafluoroisopropylidene (−C(CF3)2−) fragment in the polymer backbone which reduces the intermolecular interactions and prevents the close packing of polymer chain. The good solubility of these poly(ester-amide)s and poly(ester-imide)s can be attributed to the presence of three-dimensional triptycene units, together with the flexible segments along the polymer backbone. Therefore, the excellent solubility makes these polymers potential candidates for practical applications by simple solution casting processes. The GPC data of these polymers are listed in Table 3. All the 5′ series poly(ester-amide)s show a relatively lower inherent viscosity and molecular weight than their corresponding 5 series ones because of the lower nucleophilicity of bis(ester amine) 3′.
Thermal properties of the polymers were investigated by DSC and TGA. The thermal behavior data of these polymers are summarized in Table 4. DSC measurements were conducted with a heating rate of 20 °C/min under a nitrogen flow. Quenching from an elevated temperature of about 400 °C to 50 °C gave predominantly amorphous samples so that the glass-transition temperature (Tg) of these polymers could be easily measured in the second heating traces of DSC (Figs. 5 and 6). Tgs of poly(ester-amide)s 5a − 5e and poly(ester-imide)s 7a − 7d were observed in the range of 242–288 °C and 256–298 °C, respectively. Poly(ester-amide) 5c and poly(ester-imide) 7c displayed the lowest Tg in each series because of the presence of flexible ether linkage in their backbones. The higher Tg values associated with polymers 5b and 7a could be attributed to the presence of rigid biphenylene and pyromellitimide units that stiffen the polymer backbone.

DSC curves (the second scans after quenching from 400 °C) of the poly(ester-amide)s 5a − 5e with a heating rate of 20 °C/min in nitrogen

DSC curves (the second scans after quenching from 400 °C) of the poly(ester-imide)s 7a − 7d with a heating rate of 20 °C/min in nitrogen
The thermal stability of the polymers was evaluated by TGA in both air and nitrogen atmospheres. TGA curves of poly(ester-amide) 5a and poly(ester-imide) 7d are illustrated in Fig. 7. These polymers exhibited reasonable thermal stability without significant weight loss up to 450 °C under nitrogen or air atmosphere. The decomposition temperatures (Td) at 10% weight losses in nitrogen and air atmospheres taken from the original TGA thermograms are given in Table 4. The decomposition temperatures at 10% weight-loss of poly(ester-amide)s 5a − 5e in nitrogen and air were recorded in the range of 468–497 °C and 464–492 °C, respectively, and the poly(ester-imide)s 7a − 7d showed Td at 10% weight loss in the range of 506–516 °C and 491–504 °C, respectively. All of the polymers seemed to exhibit a two-stage decomposition behavior at elevated temperatures. The first stage of weight loss starting around 450 °C might be attributed to the early degradation of the less stable ester groups. The amount of carbonized residues (char yield) at 800 °C in nitrogen for poly(ester-amide)s 5a − 5e was in the range of 62–68 wt%, and poly(ester-imide)s 7a − 7d was in the range of 50–55 wt%. The high char yields of these polymers can be attributed to their high aromatic content.

TGA curves of the polymers: (a) 5a and (b) 7d, with a heating rate of 20 °C/min
The color intensities of the poly(ester-amide)s 5a − 5e and poly(ester-imide)s 7a − 7d were elucidated from the yellowness (b*) or redness (a*) indices observed by a colorimeter. For comparison, a standard polyimide from PMDA and ODA (the same components to the commercial Kapton film) was also prepared and characterized its color intensity. The color coordinates of all these polymer films are shown in Table 5. The slightly higher yellowness index (b*) of the thermally imidized poly(ester-imide) films might be explained by thermal oxidation of chain-end amino groups or a denser chain packing. In addition, all these polymer films were also measured for optical transparency using UV-vis spectroscopy. Figs. 8 and 9 show the UV-visible transmittance spectra of poly(ester-amide) films and some representative poly(ester-imide) films, respectively, and the cut-off wavelengths (absorption edge; λ0) from the UV-vis spectra are also listed in Table 5. All the poly(ester-amide) and poly(ester-imide) films showed cut-off wavelengths shorter than 410 nm and were entirely transparent and near colorless. All the polymer films in this work exhibited much lighter color than the standard Kapton polyimide film (see inset in Fig. 9). The low color of these poly(ester-amide)s and poly(ester-imide)s could be explained by the decreased intermolecular electronic interactions. The bulky and three-dimensional triptycene units in polyamide or polyimide component were effective in decreasing charge-transfer complexing (CTC) between the polymer chains through a steric hindrance effect.

Transmission UV-visible absorption spectra of the poly(ester-amide) films 5a − 5e with thickness of about 45 μm

Transmission UV-visible absorption spectra of the poly(ester-imide) films 7c–C, 7d–C, 7c–T and 7d–T with thickness of about 50 μm (Kapton: PMDA/ODA polyimide)
Conclusions
Two new triptycene-based bis(ester amine)s, 1,4-bis(3-aminobenzoyloxy)triptycene (3) and 1,4-bis(4-aminobenzoyloxy)triptycene (3′), have been successfully synthesized and led to new aromatic poly(ester-amide)s and poly(ester-imide)s containing triptycene moieties by polycondensation with various dicarboxylic acids and dianhydrides, respectively. Most of the polymers derived from bis(ester amine) 3 were readily soluble in polar organic solvents and could afford strong and flexible films via solution casting. The cast films showed less color intensity or colorlessness and moderate thermal stability. However, the polymers from bis(ester amine) 3′ could not afford flexible films via solution casting due to lower molecular weights.
References
Yang HH (1989) Aromatic high-strength fibers. John Wiley & Sons, Inc, New York
Sroog CE (1991). Prog Polym Sci 16:561
Ghosh MK, Mittal KL (eds) (1996) Polyimides: fundamentals and applications. Marcel Dekker, New York
Garcia JM, Garcia FC, Serna F, de la Pena JL (2010). Prog Polym Sci 35:623
Liou GS, Yen HJ (2010) Polyimides. In: Matyjaszewski K, Moller M (eds) Polymer science: A comprehensive reference, vol 5. Elsevier BV, Amsterdam, pp 497–535
Liaw DJ, Wang KL, Huang YC, Lee KR, Lai JY, Ha CS (2012). Prog Polym Sci 37:907
Imai Y (1995). High Perform Polym 7:337
de Abajo J, de la Campa JG (1999). Adv Polym Sci 140:23
Ghosh MK, Sen SK, Banejee S, Voit B (2012). RSC Adv 2:5900
Hsiao SH, Huang TL (2004). J Polym Res 11:9
Hsiao SH, Chang YH (2004). J Polym Sci Part A: Polym Chem 42:1255
Chung CL, Tzu TW, Hsiao SH (2006). J Polym Res 13:495
Mao HC, Zhang SB (2014). J Mater Chem A 2:9835
Li X, Jiang JW, Pan Y, Sheng SR, Liu XL (2015). High Perform Polym 27:37
Li CY, Yi L, Xu ST, Wu XM, Huang W, Yan DY (2017). J Polym Res 24:7
Ando S, Matsuura T, Sasaki S (1997). Polym J 29:69
Hasegawa M, Horie K (2001). Prog Polym Sci 26:259
Hasegawa M (2017). Polymers 9:520
Bartlett PD, Ryan MJ, Cohen SG (1942). J Am Chem Soc 64:2649
Skvarchenko VR, Shalaev VK, Klabunovskii EI (1974). Russ Chem Rev 43:951
Long TM, Swager TM (2003). J Am Chem Soc 125:14113
Cho YJ, Park HB (2011). Macromol Rapid Commun 32:579
Wiegand JR, Smith ZP, Liu Q, Patterson CT, Freeman BD, Guo R (2014). J Mater Chem A 2:13309
Weidman JR, Luo S, Breier JM, Buckley P, Gao P, Guo R (2017). Polymer 126:314
Zhang Q, Li S, Li W, Zhang S (2007). Polymer 48:6246
Sydlik SA, Chen Z, Swager TM (2011). Macromolecules 44:976
Hsiao SH, Wang HM, Chen WJ, Lee TM, Leu CM (2011). J Polym Sci Part A: Polym Chem 49:3109
Hsiao SH, Wang HM, Chou JS, Guo WJ, Lee TM, Leu CM, Su CW (2012). J Polym Res 19:9757
Hsiao SH, Wang HM, Chou JS, Guo WJ, Tsai TH (2012). J Polym Res 19:9902
Yamazaki N, Matsumoto M, Higashi F (1975). J Polym Sci Polym Chem Ed 13:1373
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(DOCX 1604 kb)
Rights and permissions
About this article

Cite this article
Hsiao, SH., Liao, YC. Synthesis and properties of novel organosoluble and light-colored poly(ester-amide)s and poly(ester-imide)s with triptycene moiety. J Polym Res 25, 52 (2018). https://doi.org/10.1007/s10965-018-1452-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1452-3