
Abstract
Dynamic light scattering (DLS) of polymer and polymer–nanocomposite solutions has been performed to examine the effect in the morphology of polymer solution in presence of nanoparticles analyzing their correlation functions. The size of the nanoparticle was determined using UV–Vis absorption spectroscopy measurements. Analysis of the correlation functions of polymer solution shows existence of two modes, namely, fast and slow modes, along with the distinct values in their corresponding amplitudes and relaxation times. Interestingly, the fast mode of the solution was found to smear out, enhancing the slow mode when we grow nanoparticles into the polymer solution. Apart from the above study, the temperature variation study of both the solutions show that above and below room temperature, the polymer solution becomes more heterogeneous compared to the solution when nanoparticles are grown into it.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Experimental studies of dilute, semidilute, and concentrated polymer solutions in past few decades have been investigated by Dynamic Light Scattering technique that measures the correlation functions of the scattered light intensity with certain delay times. One can extract the relaxation times that characterize the dynamics of the process from these correlation functions. Recently, many dynamic processes of various polymer–solvent systems [1, 2] and many polymer–nanoparticle systems [3,4,5,6,7,8,9] have been studied for both scientific and technical reasons.
In dilute solutions, the translational diffusion of polymer coils, internal mode of homopolymer [10], or diblock copolymer chains [11,12,13] are extensively studied. On the other hand, for both semidilute and concentrated polymer solutions, the cooperative diffusion of the transient polymer network, heterogeneity mode related to polymer self-diffusion in diblock copolymers [11,12,13,14,15], entanglement mode [16], chain reptation [16, 17], viscoelastic relaxation [18], diffusion of clusters [15, 16, 19], predicted Rouse modes theoretically [20] and observed experimentally [21], the viscoelastic relaxations, α and β - chain relaxations [18, 22, 23] are also studied. Using DLS, the chain conformation behavior as well as hydrodynamic size distribution of polymer–solvent systems have been studied by analyzing the correlation functions. In contrast, most of the existing scientific literature of polymer–nanoparticle systems have been concentrated only on the hydrodynamic size distribution. However, the study after synthesis of nanoparticles directly into the polymer solution using DLS is hardly available that has many importance in the fields of fundamental, sciences and technological applications. Nanoparticle formation into the solution will affect the dynamical behavior of polymer chain conformations, which is likely to be reflected in their correlation functions, which motivate us to perform the present study using simple DLS measurements.
Here we have prepared nanoparticles directly into an aqueous polymer solution, and the effects were examined studying the intensity autocorrelation functions obtained from dynamic light scattering measurements at room temperature. The nanoparticles were synthesized chemically by growing CdS particles into the polymer solution. In order to obtain the size of the nanoparticles, we have used the UV–Vis absorption spectroscopy technique. Comparison of the correlation functions of the polymer and polymer nanocomposite solutions shows huge change. Each correlation function of the polymer solution was found to have two modes, namely the fast and the slow modes. As we grow nanoparticles into the polymer solutions, the prominent fast mode of the polymer solution is found to smear out, increasing the slow relaxation mode by increasing both its amplitude and relaxation time. Additionally, the thermoresponsive behavior of both the solutions was studied collecting the correlation data at different temperatures. Systematic change in their correlation functions with temperature for both the solutions was observed. We found that the polymer solution is heterogeneous compared to the polymer nanocomposite solution above and below room temperature.
Experimental details
Sample preparation
The synthesis of polymer-nanocomposite is performed chemically by dispersing the CdS nanoparticles with 5% volume concentration in 5 mg/ml (in between dilute and concentrated) aqueous polyacrylamide solution having molecular weight (M w = 5 − 6 × 106). A measured amount of cadmium acetate is dissolved in 5 mg/ml polymer solution for the preparation of the polymer nanocomposite solution. Using a magnetic stirrer, the solution was stirred for the mixture of cadmium acetate homogeneously into the polymer, and H 2 S gas was then passed through the solution in a controlled way. When the gas was passed, the color of the solution become yellow ensured that the reaction of cadmium acetate with H 2 S gas occurred which in tern indicates the formation of CdS particles. The gas flow was then stopped and the solution was kept open for a few minutes to remove of excess H 2 S from the sol. The product do not show any precipitation for years which signify the fact that the composite solution is stable for long periods in water. The concentrations of polymer and polymer–nanocomposite solutions used for the study were 5 mg/ml, which is just above the overlap concentration 4 mg/ml of the polymer.
Dynamic light scattering
Using Zetasizer Nano-S system (Malvern Instruments Ltd., Malvern, UK) the dynamic light scattering measurements were performed at fixed scattering angle (173° backscatter) with a 5-ml glass cuvette. The instrument contains an avalanche photodiode (APD) detector along with a 4 mW He-Ne laser, which operates at a wavelength (λ) of 633 nm. The setup also has a Peltier temperature controller, which allows changing the temperature from 2 °C to 92 °C. The wavenumber of scattered light corresponding to the scattering angle 173° is q = (4π n/λ)sin(𝜃/2) = 0.019 nm − 1, n is the refractive index of the solution. The data are collected at room temperature as well as at various temperatures ranging from 2 to 90 °C. The temperature is controlled within ± 0.02 °C. Before each measurement at each temperature, the sample placed in the DLS apparatus is allowed 15 min to equilibrate the temperature. The data acquisition time for all dynamic light scattering measurements was 60 s per correlation function. All measurements at each temperature were performed twice to check the consistency of the correlation data. Also, the reversibility of the correlation data with temperature is checked by lowering the temperature from 90 to 2 °C. Also, it may be noted that the CdS nanocrystals are luminescent, which may contribute to the scattered light intensity. Instead of the luminescent property, DLS study of CdS nanocrystals has been performed by many authors recently [24,25,26]. The luminescent properties are well studied by many authors using photoluminescence spectroscopy technique (PL) [24,25,26,27,28,29]. The PL spectra upon excitation with a 782-nm laser showed that the PL emission profile occurs in the 480–540 nm range [28]. In our study, we measure the correlation function with a 633-nm He-Ne laser. The detector here is the avalanche photodiode detector, which has quantum efficiency greater than 50% at 633 nm. So, the detector is more efficient to collect scattered light only when the light of 633 nm (same wavelength as incident light) is incident on it, and the light of other wavelengths do not contribute more to the detected scattered light.
In DLS experiments, the intensity–intensity time correlation functions g 2(t) are recorded. If the scattered field obeys Gaussian statistics, the measured correlation function is directly related to the theoretically amenable first-order field correlation function g 1(t) through the Siegert relation [30]
where c is an instrumental parameter, a constant related to the coherence of the detection optics. For a polydisperse system [30, 31], g 1(t) is related to the distribution of the characteristic line width G(Γ) by
For a purely diffusive relaxation, Γ is related to the translational diffusion coefficient D or the cooperative diffusion coefficient D 1 by Γ = D 1 q 2, depending on whether the solution is in the dilute (C < C ∗) or semidilute (C > C ∗) regime. On the other hand, it has been shown that in a semidilute solution or a solution undergoing the cross-linking reaction, g 2(q,t) can also be analyzed by a single-exponential function combined with a stretched exponential function to take care of the additional slow relaxation as follows [32,33,34,35,36,37,38,39,40]
with A 1 + A 2 = 1. The parameters A 1 and A 2 are the amplitudes of the fast and second modes, respectively. The first term (short-time behavior) on the right-hand side of the Eq. 3 is related to the cooperative diffusion of the polymer chains. It has an amplitude A 1 and a relaxation time τ 1, which corresponds to the diffusion coefficient D 1 = (q 2 τ 1)− 1, where q is the scattering vector. For the cooperative mode of semidilute solution [41], the Stokes–Einstein relation [42] is modified as, D 1 = k B T/6π η ξ, where k B is the Boltzmann constant, η is the viscosity of the solvent at temperature T, and ξ is the hydrodynamic correlation length (correlation length of concentration fluctuation or mesh size), which is inversely proportional to polymer concentration and independent of the molecular weight [43]. Note that D 1 is a measure of the dynamics on a length scale of 1/q, which is about 50 nm at the scattering angle of 173°. The conjecture is that the second term (stretch exponential decay) on the right-hand side of Eq. 3 is associated with the disengagement relaxation of individual chains [44]. This stretch exponential decay [16, 45, 46] corresponds to the slow mode that results from the diffusion of polymer associations, known as the interdiffusive mode (which is independent of the scattering vector, characterizes the structural relaxation of the transient network) [47] is characterized by the amplitude A 2 and relaxation time τ 2. The variable τ 2 is some effective relaxation time, and β(0 < β ≤ 1) is a measure of the width of the distribution of relaxation times. Note that the stretched exponential function, in general, indicates the presence of multiple relaxation processes in disordered systems [45]. This form is known to represent well the asymptotic behavior of nonexponential relaxations of various nonequilibrium parameters of macroscopic systems. The advantage of this functional form is that it could represent the entire multirelaxations behavior of the entire autocorrelation function by only two parameters, namely, an exponent β and a correlation time τ 2. The variable τ 2 is some effective relaxation time, and β(0 < β ≤ 1) is a measure of the width of the distribution of relaxation times. The mean relaxation time is,
where Γ(1/β) is the gamma function of β − 1.
In the analysis of the correlation data, a nonlinear fitting algorithm (Levenberg–Marquardt method) has been used to obtain best-fit values of the parameters A 1,A 2, τ 1 and τ 2 using Origin 7.5 software.
UV–Vis absorption spectroscopy
The size of the CdS particles suspended in the solution is determined using UV–Vis absorption spectroscopy (Cintra 10e GBC) technique. Using transmission mode, the absorption spectra of the polymer nanocomposite solution filled in standard cuvette (10 mm × 10 mm) was taken at room temperature. In Fig. 1, the corresponding spectra is shown and in the inset the same data is plotted as α h υ 2 versus h υ. In direct band-gap semiconductors like CdS, the absorption coefficient is expressed as [48], α h υ ∝ (h υ − E g )1/2, where α is the absorbance, h is the Planck’s constant, υ is the frequency of the radiation and E g is the band gap energy. The value of h υ is extrapolated to α = 0 to obtain the band gap of CdS in polymer nanocomposite solution, which is shown in the inset of Fig. 1. It has been observed that the absorption band edge of CdS in the polymer nanocomposite solution (2.46 eV) is blue shifted [49] in the presence of nanocrystalline CdS particles compared to the band gap of bulk CdS (2.4 eV). The band gap (\({E_{g}^{p}} = 2.46\) eV) of the CdS particles in the polymer nanocomposite solution is found to be larger than the band gap of bulk CdS (\({E_{g}^{b}} = 2.4\) eV) [50, 51]. The increased band gap in CdS nanocrystals occurs due to quantum confinement effect. The difference in the band gap energies, ΔE = \({E_{g}^{p}}\) - \({E_{g}^{b}}\), employed with three-dimensional confinement model based on effective mass approximation [49] has been used to calculate the particle size, which is found to be 3.6 nm (7.2 nm diameter).

(Color online). Absorption spectra of the NC solution. The inset of the figure shows the plot of the data (α h ν)2 versus h ν. The band gap energy E g p = 2.46 eV is obtained from the extrapolation of the curve to α = 0
It is to be noted here that the calculation of size of CdS using this technique (effective mass approximation) is well verified by many other complementary techniques, like X-ray diffraction, electron microscopy, reaction with hydroxyl radicals, fluorescence quenching with methylviologen etc. [24,25,26,27,28,29, 52]. Here we have not used any such complementary techniques to verify the size of the particles because CdS is a semiconducting particle (belonging to the II-VI group) and UV–Vis measurements allow us to determine the band gap as well as the size of the particles in a more accurate way, which is mentioned by many authors as described above.
Experimental results and discussion
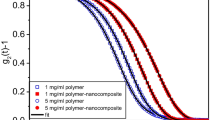
In Fig. 2, we have shown the auto correlation functions of the polymer and polymer nanocomposite solutions obtained from DLS measurements. The blue dots represent the correlation data of the polymer solution and the red dots correspond to the correlation data of the polymer nanocomposite solution. The solid lines are obtained from the fitting of the data using Eq. 1. It is clear from the figure that the correlation function for the nanocomposite solution differs widely from that of the polymer one, where CdS nano-particles are grown. It is important to note that the correlation function of the polymer solution contains two modes, namely, the fast (τ 1 − 1), and the slow (τ 2 − 1) modes, respectively. The fast mode of polymer solution corresponds to cooperative diffusion of individual chains, while the slow one might be related to the dynamics of those chains entangled in polymer solution. The parameters obtained from the fitting of the correlation functions using Eq. 1 are given in Table 1. The amplitudes of the fast and slow modes of the polymer solution are 0.49 and 0.53, and the corresponding relaxation times are 80.92 and 500.39, respectively. The amplitudes of the two modes are very close to each other and the relaxation time of the fast mode is about an order of magnitude smaller than the slow mode, while for the polymer nanocomposite solution, the amplitudes of the fast and slow modes are 0.008 and 1, and the corresponding relaxation times are 36.6 and 1275.41, respectively.

(Color online). Time correlation functions of semidilute and NC solutions. The filled blue and red circles correspond to the experimental data of polymer and NC solutions, respectively. The solid curves are obtained by fitting the data using Eq. 1
In connection with this fact, one should consider that the cooperative diffusion coefficient (fast mode) of the polymer solution is D 1 = (q 2 τ 1)− 1 = 34.23 × 10− 12 m 2/sec, which can be used to estimate the hydrodynamic screening length or mesh size following the Stokes–Einstein relation, and is found to be 7.16 nm. The mesh size of polyacrylamide in semidilute regime obtained by small-angle neutron scattering measurements is [53], ξ = 2.09c − 0.76, where c is the concentration of the solution in g/ml. We have estimated the value for our case, and the mesh size is found to be 11.72 nm. The corresponding diffusion coefficient following the Stokes–Einstein relation is found to be 20.92 × 10− 12 m 2/s, which is comparable to the estimated value as obtained using the mesh size of 7.16 nm.
It may be noted here that for polymer nanocomposite solution, the amplitude of the fast mode is negligibly smaller compared to its slow mode. The amplitude of the slow mode of polymer nanocomposite solution is also higher than the amplitude of the slow mode of the polymer solution, while the amplitude of the fast mode of polymer nanocomposite solution is very small compared to the fast mode amplitude of polymer solution. From this comparison it can be manifested that due to the presence of nanoparticles into the solution, the prominent fast mode of polymer solution is smeared out, enhancing the slow mode. This may be due to the fact that as the polymer solution is loaded with the CdS particles, the slow relaxation mode of polymer solution becomes even slower, and at the same time its contribution to the total intensity increases with the rise of its amplitude. In polymer nanocomposite material, CdS particles are formed in the aqueous solution of polymer chains, and during their growth, due to polymer-particle interaction, different parts of the polymer chains get connected with CdS particles and the individual chains are eventually cross-linked through formation of cluster. Due to both attachment of the CdS particles with polymer chains and large interfacial area-to-volume ratio of nanoparticles, the size of the cluster becomes large. As a result, the slow mode is enhanced by increasing both its amplitude and relaxation time in the correlation function. The fast mode may also present in the polymer nanocomposite solution but its contribution is suppressed due to the enhanced slow mode that results from these large clusters. Therefore, it can be predicted that the formation of nanoparticles in the polymer solution changes the morphology of the solution from the bimodal to a monomodal one, which follows the stretch exponential decay function. The uniqueness or variations of these features can be easily observed from the temperature-dependence studies of the solutions.
Apart from the above study, the thermoresponsive behavior of both the solutions was studied and DLS measurements at various temperatures were carried out. All the correlation functions are plotted in Fig. 3. Figure 3a shows the time correlation functions of the polymer solution at various temperatures. It is clear from the data that the correlation functions are strongly temperature-dependent and follow a systematic behavior with the increase or decrease in temperature. Analysis of the data has been done using Eq. 1 and the fitted curves are shown by the solid lines in the figure. Figure 3b shows the time correlation functions of the polymer nanocomposite solution at different temperatures. It has been observed from the figure that the correlation functions change systematically with temperature. The data are now analyzed only considering the second term of Eq. 3, since the polymer nanocomposite solution has only stretch exponential mode as observed from the analysis of Fig. 2, and the fitted curves are shown by the solid lines in the figure. The excellent fitness of the data with the stretch exponential function indicates that the temperature does not further enhance the original fast mode of the polymer solution. In order to check the reproducibility, the data for each correlation function at each temperature is collected repeatedly for two times. The correlation data of both the solutions are also collected with the reversal of temperature, and it is verified that for both the cases of rising and lowering of temperatures, the results match with each other.

(Color online). Time correlation functions at various temperatures. a Semidilute polymer solution and b NC solution. The lines of figure (a) are obtained by fitting the data using Eq. 1 and the lines of figure (b) are obtained by fitting the data considering only the stretch exponential decay term of Eq. 1. The temperatures are shown against the corresponding symbols
The amplitudes obtained from the fitting of the correlation functions of Fig. 3 with temperature are plotted in Fig. 4. The open and solid blue symbols represent the fast and slow modes amplitudes of polymer solution and the red symbols correspond to the slow-mode amplitude of polymer nanocomposite solution. It has been observed that for polymer solution the amplitudes of the two modes change with temperature. A crossover region between the two modes is observed around 25 °C. Above this temperature, the amplitude of the fast mode is increased, while the amplitude is decreased below this temperature compared to the slow mode. Similar opposite behavior with temperature is observed for the slow mode compared to the fast one. On the other hand, for the polymer nanocomposite solution, the amplitude remains constant. It is interesting to note that contrary to the polymer solution, the amplitude of the polymer nanocomposite solution does not change with temperature. This proves that temperature does not alter the monomodal characteristics of the polymer nanocomposite solution.

(Color online). Variation of the amplitudes of fast and slow modes for the polymer and NC solutions with temperature. The open and solid blue symbols represent the fast and slow modes amplitudes of polymer solution, respectively, and the solid red symbols correspond to the slow mode amplitudes of NC solution
The relaxation times of fast and slow modes obtained from the fitting of the correlation data (Fig. 3) with temperature are plotted in Fig. 5. The open and solid blue symbols correspond to the fast and slow modes relaxation times of polymer solution, and the solid red symbols represent the slow mode relaxation time of polymer nanocomposite solution. It has been observed that the relaxation time corresponding to the slow mode of polymer nanocomposite solution has a higher value compared to the slow mode of the polymer solution. Also, the relaxation times for both the solutions are decreased with the increase of temperature. This behavior of relaxation times with temperature might be due to the change in viscosity of the solution. It is important to note that the relaxation times of both the solutions are reversible with temperature, which reveals that the temperature does not make any permanent change to the chemical state of the solutions. Since the relaxation times are similar, we have not shown the data in the above figure.

(Color online). Variation of the relaxation times of fast and slow modes for the polymer and NC solutions with temperature. The open and solid blue symbols represent the fast and slow modes relaxation times of polymer solution, respectively, and the solid red symbols correspond to the slow mode relaxation times of NC solution
The stretching exponent β is plotted in Fig. 6 as a function of temperature. The open blue symbols represent the stretching factors of polymer solution and the solid red symbols correspond to the stretching factors of polymer nanocomposite solution. The solid lines in the figure are obtained from the linear fit to the data. It has been clearly observed from the figure that the behavior of stretching factor with temperature for polymer nanocomposite solution is totally different and follows an opposite trend compared to the polymer one. It is interesting to note here that for the polymer solution, the stretching factor shows maximum value at 25 °C, and away from this temperature the value decreases, which is clearly shown by the solid lines in the figure, while for the polymer nanocomposite solution the stretching factor shows a minimum at this particular temperature and the value increases for all other temperatures. The minimum and maximum values of stretching factor for polymer solution are 0.39 and 0.62, and that for polymer nanocomposite solution are 0.74 and 0.78, respectively. The overall change in stretching factor for both the polymer and polymer nanocomposite solutions are 37 and 5%, respectively (within the studied temperature ranges). It is interesting to note that the β value decreases 37% for polymer solution while it increases 5% for the polymer nanocomposite solution. The decrease in stretching exponent β for polymer solution indicates that the solution becomes more heterogeneous when the temperature is away from 25 °C, since the smaller value of β signifies highly stretched conformational relaxation of the polymer [44, 45]. In other words, we can say that the chains entangled in the polymer solution are not able to relax during the delay time window. This reveals that the enhancement of relaxations in the slow mode due to conformational changes of the polymer chains in the solution occurs whose decay is faster than exponential. On the other hand, the enhancement of stretching exponent β for polymer nanocomposite solution manifests that the solution is more homogeneous for these temperature ranges. Thus, it can be emphasized that the conformations of the polymer chains in the polymer nanocomposite solution is much more ordered or homogeneous with the temperature change from 25 °C. It relaxes nearly as a single exponent and at the same time the contribution of the fast diffusive relaxation of polymer solution becomes less and less since individual chains are eventually cross-linked through the attachment with CdS particles as parts of larger clusters. Hence, we can predict that with the change of temperature, the inhomogeneities in the polymer solution are largely enhanced compared to the polymer nanocomposite solution. However, the angular-dependent light scattering measurements would provide more accurate quantitative information about the heterogeneity and homogeneity behavior of the solutions. Since, in the present work we are mainly interested in showing the effect of nanoparticles in polymer solution, we have discussed qualitatively the homogeneity and heterogeneity behavior of the solutions with temperature only using the stretching parameter β for completeness of the study.

(Color online). Variation of stretching factors of polymer and NC solutions with temperature. The open blue symbols represent the stretching factors of polymer solution and the solid red symbols correspond to the stretching factors of NC solution. The solid lines are obtained from the linear fit to the data
Conclusions
The intensity autocorrelation functions of polymer and polymer nanocomposite solutions have been examined using dynamic light scattering technique. The size of the nanoparticle was estimated from the UV–Vis absorption spectroscopy measurement. The correlation functions of polymer solution show two modes namely, the fast and slow modes, and this fast mode is found to smear out enhancing the slow one when nanoparticles are grown into the solution. Additionally, the temperature variation study of both the solutions shows that the polymer nanocomposite solution is more homogeneous at above and below room temperature compared to the polymer solution.
References
Brown W (1996) In: Brown W (ed) Light scattering: principles and development, Chapter 8. Oxford Science Publications, Oxford, p 255
Brown W (1993) In: Brown w (ed) Dynamic light scattering: the method and some applications. Oxford Science Publications, Oxford, p 267
Glomm WR (2010) ACS Nano 4:1187–1201
Liang M, Lin IC, Whittaker MR, Minchin RF, Monteiro MJ, Toth I (2010) ACS Nano 4:403–413
Wu C, Niu A, Leung LM, Lam TS (1999) J Am Chem Soc 121:1954–1955
Kropka JM, Putz KW, Pryamitsyn V, Ganesan V, Green PF (2007) Macromolecules 40:5424–5432
Dong H, Zhu M, Yoon JA, Gao H, Jin R, Matyjaszewski K (2008) J Am Chem Soc 130:12852–12853
Jordan J et al (2005) Mater Sci Eng A 393:1–11
Shibayama M, Suda J, Karino T, Okabe S, Takehisa T, Haraguchi K (2004) Macromolecules 37:9606–9612
Berne BJ, Pecora R (2000) Dynamic light scattering, Chapter 8. Dover Publications, New York, p 194
Semenov AN, Fytas G, Anastasiadis SH (1994) Polym Prepr (Am Chem Soc, DiV Polym Chem) 35:618
Pan C, Maurer W, Lu Z, Lodge TP, Stepanek P, von Meerwal ED, Watanabe H (1995) Macromolecules 28:1643–1653
Jian T, Anastasiadis SH, Semenov AN, Fytas G, Adachi K, Kotaka T (1994) Macromolecules 27:4762–4773
Stepanek P, Tuzar Z, Kadlec P, Kriz J (2007) Macromolecules 40:2165–2171
Borsali R, Giebel L, Fischer EW, Meier G (1991) J Non-Cryst Solids 131:816–822
Stepanek P, Brown W (1998) Macromolecules 31:1889–1897
Adam M, Delsanti M (1985) Macromolecules 18:1760–1770
Brown W (1996) In: Brown W (ed) Light scattering: principles and development, Chapter 11. Oxford Science Publications, Oxford, p 352
Stepanek P, Lodge TP (1996) Macromolecules 29:1244–1251
Semenov AN (1990) Physica A 166:263–287
Boudenne N, Anastasiadis SH, Fytas G, Xenidou M, Hadjichristidis N, Semenov AN, Fleischer G (1996) Phys Rev Lett 77:506–509
Floudas G, Placke P, Stepanek P, Brown W, Fytas G, Ngai K (1995) Macromolecules 28:6799–6807
Floudas G, Stepanek P (1998) Macromolecules 31:6951–6957
Ipe BI, Shukla A, Lu H, Zou B, Rehage H, Niemeyer CM (2006) ChemPhysChem 7:1112–1118
Limin Q, Helmut C, Markus A (2001) Nano Lett 1:61–65
Baykul MC, Rzgar H, Arman E, Ba Y, Btn V (2010) Phys Status Solidi C 7:423–426
Joseph RL, Ignacy G, Zygmunt G, Catherine JMJ (1999) Phys Chem B 103:7613–7620
Pushan A, Parinda V, Praveen T (2005) J Appl Phys 97:104310
Paola F, Depalo N, Comparelli R, Curri ML, Striccoli M, Castagnoloand M, Agostiano1 A (2008) J Therm Anal Calorim 92:271–277
Berne BJ, Pecora R (1976) Dynamic light scattering. Wiley, New York
Chu B (1991) Laser light scattering, 2nd edn. Academic Press, New York
Kohlrausch R (1847) Phys Pogg Ann Chem 72:353
Williams G, Watts DC (1970) Trans Faraday Soc 66:80–85
Ngai T, Chi W (2003) Macromolecules 36:848–854
Ngai T, Chi W, Chen Y (2004) Macromolecules 37:987–993
Chen H, Ye XI, Zhang G, Zhang Q (2006) Polymer 47:8367–8373
Shibayama M, Norisuye T (2002) Bull Chem Soc Jpn 75:641–659
Adam M, Delsanti M, Munch JP, Durand D (1988) Phys Rev Lett 61:706–709
Martin JE, Wilcoxon J (1988) Phys Rev Lett 61:373–376
Martin JE, Wilcoxon J, Odinek J (1991) Phys Rev A 43:858–872
Adam M, Delsanti M (1977) Macromolecules 10:1229–1237
Doi M, Edwards SF (1986) The theory of polymer dynamics, Chapter 3. Clarendon Press, Oxford, p 49
Nicolai T, Brown W, Johnsen RM, Stepanek P (1990) Macromolecules 23:1165–1174
Douglas JF, Hubbard JB (1991) Macromolecules 24:3163–3177
Phillips JC (1996) Rep Prog Phys 59:1133–1207
Huber DL (1985) Phys Rev B 31:6070–6071
Hong PD, Chou CM, Chen JH (2000) Polymer 41:5847–5854
Kang J, Tsunekawa S, Kasuya A (2001) Appl Surf Sci 174:306–309
Wang Y, Heron N (1990) Phys Rev B 42:7253–7255
Nanda KK, Sarangi SN, Sahu SN (1997) Curr Sci 72:110–111
Soloviev VN, Eichhofer A, Fenske D, Banin U (2000) J Am Chem Soc 122:2673–2674
Weller H, Schmidt HM, Koch U, Fojtik A, Baral S, Henglein A (1986) Chem Phys Lett 124:557–560
Wu C, Quesada MA, Schneider DK, Farinato R, Studier FW, Chu B (1996) Electrophoresis 17:1103
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mondal, M.H. Study of autocorrelation function of polymer and polymer–nanocomposite solutions using dynamic light scattering method. J Polym Res 24, 218 (2017). https://doi.org/10.1007/s10965-017-1368-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-017-1368-3