
Abstract
Polyalkylthiophenes are applied in the form of thin films as active layers in organic devices. The main properties defined in the synthetic procedure that can affect the film formation are the molar mass and regioregularity degree (the content of head-to-tail, HT, linkages). These properties can be varied, at principle, by changing the conditions of the oxidative polymerization process. In this work, we evaluate the effect of oxidant addition rate, temperature and time-dependence of poly-(3-hexylthiophene), P3HT, oxidative polymerization in molar mass and regioregularity degree, besides other polymer properties such as absorption and emission of visible light. The results show that the polymer started to grow during the oxidant addition and already presents a relatively high molar mass (ca. 10,000 g/mol) just after the addition stopped. Polymerization temperature is more significant in molar mass variations than the time expended for the polymerization reaction, with values of Mw ranging from 15,000 to 70,000 g/mol in the conditions tested. The HT contents were all above 70 %, with higher variations in the two first hours of polymerization reaction and are mainly defined during the oxidant addition, which leads to higher HT contents and narrower molar mass distributions using slower additions. The solvent extraction reveals that the HT content is directly related to the polymer chains extension, being possible to improve both regioregularity degree and molar mass of P3HT.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polythiophenes present higher thermal and chemical stabilities and easiness of side-chain functionalization [1] comparing to other conjugated polymers, allowing the preparation of several derivatives with improved physical properties, including electroluminescent and conductivity, for the development of several devices, such as, organic light emitting diodes, transistors, photo-voltaic cells and solid state sensors [2, 3]. The repeating units can be linked in the main-chain in three ways: head-to-tail (HT), tail-to-tail (TT) or head-to-head (HH) leading to different properties. High content of HT linkages, which are more regioregular, present high crystallinity and conductivity [4]. Despite several new conjugated polymers are being applied for applications on photovoltaics devices [5] or on field-effect transistors [6] with claimed better performance and efficiency, polythiophene derivatives, especially, the poly-(3-hexylthiophene) (P3HT) is still a standard in this field and probably, one of the most studied conjugated polymer. Moreover, technological studies for mass production of organic devices are usually done with P3HT [7–9]. The main polythiophenes applications are as active layers in devices and sensors, usually as thin film, therefore the quality and the morphology of the polythiophene films influence the application performance [10–13]. Factors such as molar mass and the degree of HT linkages are important issues on polymer film formation and for defining their morphology. Besides, for optical sensor applications, e.g. for volatile organic compounds, the regioregularity affects the response, more regioregular being less sensitive [14].
There are several synthetic routes to obtain polythiophene derivatives, which were covered on two recent reviews. Metal assisted coupling reactions, include the organometallic routes yield highly regioregular polymers [2, 15, 16], however, the reactants used are moisture sensitive, have several steps being time consuming, and expensive. McCullough and Rieke methods along with Grignard methathesis (GRIM) are the most studied methods, being Rieke the most used in commercial small scales [2, 17–19]. McCullough and GRIM are based on the polymerization of Grignard-2-halothiophene monomers using diphenylphosphino propane-nickel complex as catalyst [20]. Rieke introduced the highly reactive metal powders for preparation of Grignard compounds [21] and the use of Rieke Zinc and 2,5-dihalothiophenes in cryogenic conditions with nickel complex catalyst to prepare highly regioregular poly(alkylthiophenes) [18]. Another method for preparing regioregular polythiophenes is the Stille-protocol, based on polymerization reaction of dihalo and bis(trialkylstannyl)-funcionalized monomers using palladium complexes as catalysts. Although high regioregularity and molecular weight with good yields can be achieved, some drawbacks due to the formation of amounts of toxic species and possible instability of organometallic reagents are also present [22]. The Suzuki polymerization typically involves the use of dihalo and bis(organoboron)-funcionalized monomers [23] in a laborious protocol multiphase reaction which may present higher difficulty to control, but is largely used for preparation of copolymers, terpolymer and higher combinations of different monomers such as benzothiazoles, bothiophenes and fluorenes [23]. In recent years, the direct (hetero)arylation polycondensation was applied as alternative methodology for regioregular P3HT polymerization using 2-halo-thiophenes and heterogeneous palladium catalysts that is proceeded in fewer reaction steps and generate reduced waste [19]. It achieves high molecular weight with low polydispersity and regioregularity up to 98 % but the regioregularity is only achieved with rings having preferential CH reactive positions [24], needing high temperatures (i.e. 120 °C) and long reaction times (over two days). This method is still under intense research focused on optimization and broadening of its application for the preparation several different polymers and still have some drawbacks for large scale production [25].
The oxidative polymerization with ferric chloride, on the other hand, is a one-pot, cheap and easy process to obtain polythiophenes in large scales [19, 26–28], since do not requires leaving groups or removal of byproducts [19]. It was first suggested by Sugimoto et al. in 1986 [28], the use of monomer and ferric chloride at a 1:4 mol proportion in chloroform for polythiophene production. The method was further explored by several researchers for production of polythiophenes with different functional groups on the side chains such as alkoxy, hydroxy, azo etc. and their random copolymers [29–32]. The knowledge of how some conditions affects the oxidative polymerization process and the final polymer properties, can turn this simple procedure more useful for tuning the polymer properties. The synthetic procedure defines the molar mass and regioregularity of the polymers and in oxidative polymerization these properties can, in principle, be modulated by the experimental conditions used. For example, polymerizations at low temperatures and slow addition of oxidant lead to poly(3-octylthiophene) with regioregularities up to 80 % and relatively low molar mass polydispersity [33]; while reactions at room temperature and quick addition of solid oxidant lead to poly(3-alkyllthiophene)s with regioregularities around 65 % and broad molar mass distributions [34]. The use of low temperatures (−45 °C) in conjunction with low concentrations and long reaction times (72 h) led to polymers with 91 % regioregularity with high molecular weight and good yields [27], but the long time at too low temperature turn hard the application of the method for large scale production. Andersson et al. [26] showed that the use of a voluminous group on the side-chain of the thiophene monomer and a slow addition of the oxidant could lead to highly regioregular polymer but the contribution of the steric hindrance produced by the side-chain and the rate of oxidant addition to the regioregularity could not be separated.
The aim of this paper is to establish whether simple variations applied to the poly(3-hexylthiophene) oxidative polymerization method can lead to different molar masses and regioregularities and elucidate the influence of the reaction parameters on them. Aiming at possible large-scale production, relatively short reaction times and moderated low temperatures were explored along with different oxidant addition rates. The modulation of molar mass and regioregularity is fundamental to the development of new applications that uses polythiophenes as the active layer, such as optical sensors.
Materials
3-Hexylthiophene, poly-(3-hexylthiophene) regiorandom (ca. 50 % HT) and regioregular (ca. 98 % HT) and anhydrous ferric chloride were purchased from Aldrich and used without further purification. Chloroform and nitromethane (J. T. Baker) were dried over molecular sieve 4 Å.
Synthesis
The oxidative polymerizations were performed under nitrogen flow in a jacket glass reactor with a five-necked lid coupled to an addition funnel, a septum for sample removal, a thermometer and inlet/outlet needles for dry nitrogen flow. A circulating thermostatic bath of water/ethylene glycol kept the temperature constant. Anhydrous ferric chloride (12 mmol) dissolved in dry nitromethane (6 mL) was added dropwise onto 3 mmol of monomer dissolved in 35 mL of dry chloroform under stirring. The samples were precipitated in ethanol, filtered, washed successively with ethanol, water, 1 M ammonium hydroxide, water (until neutral pH) and ethanol and the black powders dried in a vacuum oven at 40 °C for at least 12 h. All samples were completely soluble in common organic solvents such as chloroform, tetrahydrofuran (THF) and dichlorobenzene.
Effects of polymerization time
After the addition time of the oxidant of 7 min, samples of ca. 2 mL were collected through the septum at one-hour intervals until the reaction completes six hours, counting the zero-time sample immediately after the oxidant addition stopped.
Effect of oxidant addition time
The oxidant solution was added to the reactor, either through the addition funnel or through using a peristaltic pump, during 7, 17 and, 180 min (ca. 1.7; 0.7 and 0.07 mmol/min).
Effect of reaction temperature
The circulating bath was set about 5 degrees lower than the reactor temperature, which were +10, 0, −10 and −20 °C. Reactions made at higher temperatures yielded insoluble fractions and therefore were not analyzed.
Soxhlet extraction
After completely dried, the P3HT was submitted to a soxhlet extraction with n-hexane and acetone. The process stopped when the solvent at the extraction chamber appears colorless. The insoluble powder fraction was dried as before and the soluble fraction was recovered by removing the solvent with a rotary evaporator and then dried in a vacuum in the same conditions.
Structural characterization
Molar Mass Distributions (MMD) were determined by High Performance Size Exclusion Chromatography (HPSEC) in an Agilent 1100 chromatography system with a refractive index detector, using 4 Styragel columns (500, 103, 104 and 105 Å), polystyrene standards and THF as solvent at 1 ml/min and 35 °C. 1H NMR spectra were recorded in a Bruker 400 MHz with CDCl3 as solvent and TMS as reference. The absorption and emission spectra of polymer solutions in chloroform (0.05 mg/mL) were measured in a spectrophotometer (HITACHI U-2900) and in a spectrofluorophotometer (SHIMADZU RF-5301PC) respectively. The excitation wavelength was 430 nm and excitation and emission slits, 3 nm. The Fourier Transform Infrared spectra were recorded from films, cast from CHCl3 solutions on NaCl windows, in a FTIR spectrometer (Nicolet Nexus 470 FT-IR) in transmission mode.
Results and discussions
Since the volume or the mass extracted from the reactor could not be controlled with accuracy, due to the volatility of the solvents and monomer, the quantification of non-reacted monomer and the polymer yields were not performed for reactions with aliquots taken. For the other samples, the yield were around 72–76 %. All samples of P3HT obtained were completely soluble in chloroform, toluene, xylenes, 1,2-dichlorobenzene and THF even in concentrations up to 20 mg mL−1. Both polymerization time and temperature influenced on the time spent for dissolving the samples: samples prepared with higher polymerization times and temperatures required several minutes to solubilize completely while those prepared at lower temperatures are readily dissolved.
The MMD profiles of all samples can be found in Fig. S1 in Supporting Information. The sample taken just after the addition of oxidant stopped (0 h) comprises polymer chains of significant size at all reaction temperatures, which means the polymerization already occurs during the oxidant addition [31, 33]. Moreover, the MMD peak increases exponentially with the temperature for the 0 h samples (Fig. S2), indicating that the temperature during the oxidant addition is an important factor for the chain growing.
In all cases, after the first hour of reaction, the MMD peaks shift just slightly towards higher values, being all in the range of 15,000–35,000 g/mol, but the trends with temperatures are not as clear as for sample zero. However, the MMD widths increase with the reaction time, more significantly at lower temperatures, resulting in different average molar masses as showed in Fig. 1. Thus, the temperature is more critical in the oxidant addition step and in the first hour of reaction than in the rest of the polymerization process for modulating the molar mass (Fig. 1a, b).

Evolution of molar mass averages by (a) weight (Mw); (b) number (Mn); (c) polydispersity upon the polymerization time, with an oxidant addition time of 7 min. The lines are only to guide the eyes and the bars represent the standard error
The molar mass average by weight, Mw, has more influence of the polymer chains with higher molar mass. In our case (Fig. 1a), the evolution of Mw with the polymerization time is crescent in all temperatures studied, although more accentuated at 10 °C. In contrast, the values of molar mass average by number, Mn, which has more influence of the polymer chains with lower molar mass, grows with the polymerization time just slightly in all temperatures, being practically constant after 1–2 h of reaction (Fig. 1b). This difference in the evolution of Mw and Mw values reflects on the polydispersities indexes exhibited on Fig. 1c, in which larger values of polydispersities are shown for samples obtained at the higher temperature. All the Mn, Mw and PDI values could be followed through the Tables S1-S4 in Supporting Information.
The growth of the polymer chain upon changes in the rate of oxidant addition was explored at 0 °C and three hours of reaction. The comparison of MMD profile of these syntheses is shown in the Fig. 2a. Worth of note is the profile of the sample from the synthesis with 180 min of oxidant addition, which shows a significant shift of the MMD peak to higher values of molar mass and a narrowing of the distribution profile. It seems that the amount of oxidized species in the system, produced during the oxidant addition, has a pronounced influence on the size and polydispersity of the chains.

Molar mass distributions (MMD) profiles for samples produced with different oxidant addition rates
Since the higher molar masses were achieved at 10 °C and longer oxidant addition times, another polymerization in “optimized” conditions was carried out at 10 °C and the oxidation addition of 360 min. The reaction was stopped just after the oxidation addition stopped. The MMD, compared to a sample obtained in similar condition with the quicker oxidant addition is shown in Fig. 2b.
For 360 min of oxidant addition at 10 °C (PDI 3.39, Mw 295,700 and Mn 87,300) the MMD profile reveals practically no chains with molecular weight below 104 g mol−1 (Fig. 2b). At this point, we can consider a predominant influence of faster oxidant addition rate and higher polymerization temperatures on increasing the polydispersity index, whereas an expressive increase on chain length is produced by slow oxidant addition.
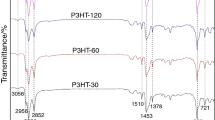
FTIR spectra of the polymers were recorded, in the form of thin films, to probe if changes on the functional groups are introduced in the polymer chain according to the reaction conditions. The great similarity of all spectra indicates that significant changes did not take place. All these features can be followed through the Fig. S3 in Supporting Information. In brief, the absence of a band in the region from 750 to 780 cm−1 characteristic of the C-Hα deformation out-of-plane in monosubstituted thiophene rings indicated that there is no residual monomer in the samples that could be determined by FTIR [35]. The symmetric deformation of methyl groups appears at 1380 cm−1 and the C = C stretching band of thiophene rings at 1510 and 1560 cm−1, methyl and methylene symmetric and asymmetric stretching bands appear at 2850, 2870, 2920 and 2950 cm−1. The absorption associated to the aromatic C-H stretching can be seen at around 3050 cm−1. The methyl asymmetric deformation band appears at 1456 cm−1 overlapped by the methylene scissor vibration at around 1465 cm−1. Aiming at access information on the regioregularity of the samples by FTIR measurements, the spectra of commercial P3HT regioregular (ca. 98 % HT) and regiorandom (ca. 50 % HT) were recorded. Worth of note are the variations of the C = C stretching wavenumber (1510 for the regioregular and 1515 cm−1 for the regiorandom), of the CH deformation (820 cm−1 for the regioregular and 828 cm−1 for the regiorandom) and the relative intensity of 1456 cm−1 and 1465 cm−1 bands, that can be a qualitative indicative of the regioregularity. There are other small differences between the regioregular and the regiorandom polymers spectra, mainly in the fingerprint region that are difficult to attribute but can also be related to residual doping [36]. The FTIR spectra of all P3HT samples obtained in this work resembles more that of the regioregular polymer than the regiorandom one with clear trends on the relative intensity 1456 cm−1 and 1465 cm−1 bands, but this analysis could be used only for qualitative assessments on the regioregularity. Attempts on quantitative determination by FTIR on films resulted in poor correlations with both relative areas and peak heights, probably due to lack of homogeneity on the sample thicknesses.
For a quantitative determination of the regioregularity degrees [37], 1H NMR spectra were recorded for all samples in CDCl3 solutions The representative 1H NMR spectra can be found the Fig. S4 in Supporting Information. A single peak at around 6.9 ppm is attributed to the aromatic proton at position 4 of the thiophene ring. The peaks appearing between 0.5 and 2 ppm are attributed to the methyl and four of the methylene groups of the side-chain. The signal of methylene linked to thiophene ring is affected by the regioregularity of the polymer. The methylene signal of the dyad HT appear at 2.79 ppm and the methylene of the HH or TT dyad at 2.56 ppm. Therefore, the regioregularity degree was calculated by the ratio of these two peaks area. Figure 3a shows that regioregularity degrees for all samples are above 69 % and it increases slightly with the reaction time, the more substantial increase occurs in the initial 2 h of polymerization. However, there is no clear trend of the regioregularity degree with the reaction temperature, in the range studied here. Therefore, the regioregularity seem to be induced during the addition of the oxidant and the reaction temperature is not the main factor in the regioregularity definition.

(a) Regioregularity degree of the samples prepared in four different polymerization temperatures; (b) Variation of the wavelength of maximum absorption with the polymerization time; (c) Variation of the Stokes’ shift with the polymerization time. The lines are only to guide the eyes and the bars represent the standard error
The influence of the polymerization time and temperature, for a fixed oxidant addition time, on the absorption and emission properties of chloroform solutions of P3HT can be analyzed from Fig. 3b, c. The wavelength of maximum emission for all samples falls in the range of 570–572 mm while the wavelength of maximum absorption (λmax) varied from 427 to 440 nm (Fig. 3b), therefore different Stokes’ shift were obtained (Fig. 3c). The λmax values increased in the first 1–2 h of polymerization then remain almost constant for all temperatures.
The dependence of the λmax with the regioregularity degree was evaluated and is shown on Fig. 4. A linear tendency of the λmax on the regioregularity is visible, although with some fluctuations. This is because even small differences in the doping level can lead to changes in the λmax that are difficult to detect by the other techniques used. With the slower oxidant addition, the range of λmax is higher those obtained with faster of oxidant addition (e.g. 434–440 for 17 min and 430–435 for 7 min) at the same reaction temperature. The amount of oxidized species produced through a slight slow addition of oxidant at the initial polymerization steps can directly influence the polymer structure. All characterization results can be followed through the Tables S1-S5 in Supporting Information.

Relationship between the wavelength of maximum absorption and the regioregularity degree of the samples. The bars represent the standard error
Solvent extraction
The low solubility of P3HT in solvents such as n-hexane and acetone allow the extraction of low molecular weight compounds and thus the evaluation the contribution of soluble and insoluble fractions in these solvents to the polymer regioregularity [27]. The extraction was performed using a soxhlet to separate the smaller fractions from the larger chains. Three synthesized samples with different MMD, polydispersity index and regioregularity degree was chosen and the results are shown in Table 1. In all cases, the regioregularity degree of the pristine samples slight increases in the insoluble fraction and showed a significant decrease in the soluble fraction, due the extraction process. The lower the regioregularity degree of the soluble fraction, the greater is the difference between the pristine and insoluble fraction. The pristine sample carried with 360 min of oxidant addition was almost completely insoluble in acetone. In this case, the fraction least regioregular was extracted in the first extraction step with n-hexane, confirmed by the second extraction with acetone lead to approximately the same degree of regioregularity. No significant differences in extraction efficiency were observed between these two solvents.
The results showed in Fig. 5 suggest a possible direct correlation between the MMD and regioregularity. As highlighted in each frame (Fig. 5b, d), P3HT chains preferably smaller than 104 g mol−1 were extracted by the solvents used. The similarity of both insoluble fractions distributions shown on Fig. 5b is a result of the extent of the first extraction with n-hexane, which extracted almost all the soluble fraction. It is observed that acetone has poor solubility power compared to n-hexane and could extract chains with size preferably below 5.103 g mol−1 (Fig. 5d), which also showed the lowest regioregularity degree, 42,8 % (Fig. 5c).

Relation between MMD and regioregularity of the samples submitted to soxhlet extraction. The methylene signal of the diad HT (2.29 ppm) and HH or TT (2.56 ppm), (a) and (c); corresponding shift of MMD, (b) and (d). All related to the samples before and after the extraction process
Conclusions
In the oxidative polymerization of the 3-hexylthiophene, our results showed that the polymer is formed during the addition of oxidation, and as soon the addition stopped, molar masses around 10,000 g/mol as denoted by the peak of the molar mass distributions are obtained. The regioregularity degree increase with the polymerization time, ca 6 % in 6 h, but it is determined mainly during the oxidant addition. The temperature of polymerization takes an important role in the chain growth, with lower molar masses and smaller polydispersities obtained at lower temperatures. Therefore, the rate or mode of oxidant addition, with the proper choice of reaction temperature, seems to be the crucial step in this kind of polymerization process to obtain products with the desired molar mass and regioregularity degree and, probably, a longer oxidant addition than tried here can lead to further increase on the regioregularity degree. It was possible to improve the regioregularity by extracting the low molecular weight fraction, n-hexane being effective to extract chains less than 104 g mol−1, and acetone preferably up to 5.103 g mol−1, showing that shorter chains are associated with lesser regioregularity. Taking into account the easiness of the procedure and the facile high scale production provided by the method, besides the possibility of improve the regioregularity a little further, the use of these polymer in electrical-optical devices such as optical sensor is promising, since the high crystallinity provided by the almost 100%HT content in highly regioregular polymers can be prejudicial to the response of this kind of sensor.
References
McCullough RD, Ewbank PC (1997) Regioregular, head-to-tail coupled poly(3-alkylthiophene) and its derivatives. In: Skotheim TA, Reynolds J (eds) Handb. Conduct. Polym. Vol. 1 Conjug. Polym. Theory, Synth. Prop. Charact., 3rd ed. CRC Press, Boca Raton, pp 225–258
McCullough RD (1998) The Chemistry of Conducting Polythiophenes. Adv Mater 10:93–116. doi:10.1002/(SICI)1521-4095(199801)10:2<93::AID-ADMA93>3.0.CO;2-F
Perepichka IF, Perepichka DF, Meng H, Wudl F (2005) Light-Emitting Polythiophenes. Adv Mater 17:2281–2305. doi:10.1002/adma.200500461
Roncali J (1997) Synthetic Principles for Bandgap Control in Linear π-Conjugated Systems. Chem Rev 97:173–206. doi:10.1021/cr950257t
He Z, Zhong C, Su S, et al. (2012) Enhanced power-conversion efficiency in polymer solar cells using an inverted device structure. Nat Photonics 6:593–597. doi:10.1038/nphoton.2012.190
Hu Y, Warwick C, Sou A, et al. (2014) Fabrication of ultra-flexible, ultra-thin organic field-effect transistors and circuits by a peeling-off method. J Mater Chem C 2:1260–1263. doi:10.1039/C3TC31869A
Søndergaard R, Manceau M, Jørgensen M, Krebs FC (2012) New Low-Bandgap Materials with Good Stabilities and Efficiencies Comparable to P3HT in R2R-Coated Solar Cells. Adv Energy Mater 2:415–418. doi:10.1002/aenm.201100517
Angmo D, Dam HF, Andersen TR, et al. (2014) All-Solution-Processed, Ambient Method for ITO-Free, Roll-Coated Tandem Polymer Solar Cells using Solution-Processed Metal Films. Energy Technol 2:651–659. doi:10.1002/ente.201402012
Sankaran S, Glaser K, Gärtner S, et al. (2016) Fabrication of polymer solar cells from organic nanoparticle dispersions by doctor blading or ink-jet printing. Org Electron 28:118–122. doi:10.1016/j.orgel.2015.10.011
Nguyen T-Q, Martini IB, Liu J, Schwartz BJ (2000) Controlling Interchain Interactions in Conjugated Polymers: The Effects of Chain Morphology on Exciton − Exciton Annihilation and Aggregation in MEH − PPV Films. J Phys Chem B 104:237–255. doi:10.1021/jp993190c
Shi Y, Liu J, Yang Y (2000) Device performance and polymer morphology in polymer light emitting diodes: The control of thin film morphology and device quantum efficiency. J Appl Phys 87:4254. doi:10.1063/1.373062
Bradley DDC, Grell M, Grice A, et al. (1998) Polymer light emission: control of properties through chemical structure and morphology. Opt Mater (Amst) 9:1–11. doi:10.1016/S0925-3467(97)00067-0
Szarko JM, Guo J, Liang Y, et al. (2010) When Function Follows Form: Effects of Donor Copolymer Side Chains on Film Morphology and BHJ Solar Cell Performance. Adv Mater 22:5468–5472. doi:10.1002/adma.201002687
Sanfelice RC (2014) Incorporação de nanopartículas metálicas a polímeros conjugados. Preparação, caracterização e utilização na fabricação de filmes nanoestruturados. Universidade de Sao Paulo
Chen T-A, Lee T-J, Lin M-Y, et al. (2010) Regiocontrolled Synthesis of Ethene-Bridged para -Phenylene Oligomers Based on Pt II - and Ru II -Catalyzed Aromatization. Chem - A Eur J 16:1826–1833. doi:10.1002/chem.200902231
Loewe RS, Khersonsky SM, McCullough RD (1999) A Simple Method to Prepare Head-to-Tail Coupled, Regioregular Poly(3-alkylthiophenes) Using Grignard Metathesis. Adv Mater 11:250–253. doi:10.1002/(SICI)1521-4095(199903)11:3<250::AID-ADMA250>3.0.CO;2-J
McCullough RD, Lowe RD, Jayaraman M, Anderson DL (1993) Design, synthesis, and control of conducting polymer architectures: structurally homogeneous poly(3-alkylthiophenes). J Org Chem 58:904–912. doi:10.1021/jo00056a024
Chen T-A, Wu X, Rieke RD (1995) Regiocontrolled Synthesis of Poly(3-alkylthiophenes) Mediated by Rieke Zinc: Their Characterization and Solid-State Properties. J Am Chem Soc 117:233–244. doi:10.1021/ja00106a027
Marrocchi A, Lanari D, Facchetti A, Vaccaro L (2012) Poly(3-hexylthiophene): synthetic methodologies and properties in bulk heterojunction solar cells. Energy Environ Sci 5:8457. doi:10.1039/c2ee22129b
McCullogh RD, Williams SP, Tristam-Nagle S, et al. (1995) The first synthesis and new properties of regioregular, head-to-tail coupled polythiophenes. Synth Met 69:279–282. doi:10.1016/0379-6779(94)02449-9
Rieke RD (1989) Preparation of Organometallic Compounds from Highly Reactive Metal Powders. Science 246(80-):1260–1264. doi:10.1126/science.246.4935.1260
Marrocchi A, Facchetti A, Lanari D, et al. (2016) Current methodologies for a sustainable approach to π-conjugated organic semiconductors. Energy Environ Sci 9:763–786. doi:10.1039/C5EE03727A
Suzuki A (1999) Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J Organomet Chem 576:147–168. doi:10.1016/S0022-328X(98)01055-9
Wang Q, Takita R, Kikuzaki Y, Ozawa F (2010) Palladium-Catalyzed Dehydrohalogenative Polycondensation of 2-Bromo-3-hexylthiophene: An Efficient Approach to Head-to-Tail Poly(3-hexylthiophene). J Am Chem Soc 132:11420–11421. doi:10.1021/ja105767z
Hayashi S, Kojima Y, Koizumi T (2015) Highly regioselective Pd/C-catalyzed direct arylation toward thiophene-based π-conjugated polymers. Polym Chem 6:881–885. doi:10.1039/C4PY01426J
Andersson MR, Selse D, Berggren M, et al. (1994) Regioselective polymerization of 3-(4-octylphenyl)thiophene with FeCl3. Macromolecules 27:6503–6506. doi:10.1021/ma00100a039
Amou S, Haba O, Shirato K, et al. (1999) Head-to-tail regioregularity of poly(3-hexylthiophene) in oxidative coupling polymerization with FeCl3. J Polym Sci Part A Polym Chem 37:1943–1948. doi:10.1002/(SICI)1099-0518(19990701)37:13<1943::AID-POLA7>3.0.CO;2-X
Yoshino K, Hayashi S, Sugimoto R (1984) Preparation and Properties of Conducting Heterocyclic Polymer Films by Chemical Method. Jpn J Appl Phys 23:L899–L900. doi:10.1143/JJAP.23.L899
Zagórska M, Kulszewicz-Bajer I, Proń A, et al. (1998) Preparation and Spectroscopic and Spectroelectrochemical Characterization of Copolymers of 3-Alkylthiophenes and Thiophene Functionalized with an Azo Chromophore. Macromolecules 31:9146–9153. doi:10.1021/ma9806561
Costa Bizzarri P, Andreani F, Della Casa C, et al. (1995) Ester-functionalized poly(3-alkylthienylene)s: substituent effects on the polymerization with FeCl3. Synth Met 75:141–147. doi:10.1016/0379-6779(95)03401-5
Hu X, Xu L (2000) Structure and properties of 3-alkoxy substituted polythiophene synthesized at low temperature. Polymer (Guildf) 41:9147–9154. doi:10.1016/S0032-3861(00)00299-8
Leclere P, Surin M, Brocorens P, et al. (2006) Supramolecular assembly of conjugated polymers: From molecular engineering to solid-state properties. Mater Sci Eng R Reports 55:1–56. doi:10.1016/j.mser.2006.12.001
Torres BBM, Balogh DT (2012) Regioregular improvement on the oxidative polymerization of poly-3-octylthiophenes by slow addition of oxidant at low temperature. J Appl Polym Sci 124:3222–3228. doi:10.1002/app.35441
Gonçalves VC, Costa LMM, Cardoso MR, et al. (2009) Synthesis and characterization of copolymers of alkyl- and azo-thiophenes: Chromic properties and photoinduced birefringence. J Appl Polym Sci 114:680–687. doi:10.1002/app.30575
Nicho M (2004) Synthesis of derivatives of polythiophene and their application in an electrochromic device. Sol Energy Mater Sol Cells 82:105–118. doi:10.1016/j.solmat.2004.01.009
Hayes W, Pratt FL, Wong KS, et al. (1985) Absorption of doped polythiophene in the middle and far infrared. J Phys C Solid State Phys 18:L555
Barbarella G, Bongini A, Zambianchi M (1994) Regiochemistry and Conformation of Poly(3-hexylthiophene) via the Synthesis and the Spectroscopic Characterization of the Model Configurational Triads. Macromolecules 27:3039–3045. doi:10.1021/ma00089a022
Acknowledgments
FAPESP, CNPq, INCT-INEO.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 309 kb)
Rights and permissions
About this article

Cite this article
Facchinatto, W.M., Torres, B.B.M. & Balogh, D.T. One-pot synthesis of poly-(3-hexylthiophene) with variable degrees of molar mass and regioregularity. J Polym Res 23, 187 (2016). https://doi.org/10.1007/s10965-016-1087-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-016-1087-1