
Abstract
The crystallization behavior and morphological structure of isothermally melt-crystallized and cold-crystallized poly(ethylene/trimethylene terephthalate) (ET) copolyesters were investigated by differential scanning calorimetry, wide-angle X-ray diffraction, and small-angle X-ray scattering. At a given crystallization temperature, the rate of cold crystallization was found to be faster than that of melt crystallization. The Avrami analysis revealed that the nuclei formed rapidly during the cooling process. This suggests that the nuclei formed during rapid cooling and that the athermal nucleation mechanism contributes to the cold-crystallization process of ET copolyesters. The crystal thickness (l c ), amorphous layer thickness (l a ), and long period (L) of ET53 and ET82 between the isothermal melt-crystallization and cold-crystallization had no significant difference. The values of the linear crystallinity (ϕ c lin) for PET, PTT, and ET copolyesters under isothermal melt-crystallization and cold-crystallization conditions were similar. This means that the random distribution of the constitutional repeating unit in the molecular chain of ET copolyesters was not exclusively in the lamella formation.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Much research has been done on the crystallization and melting behaviors of poly(ethylene terephthalate) (PET) and poly(trimethylene terephthalate) (PTT) homopolymers [1–7], which became textbook examples [8]. The chain flexibility of PTT is higher than that of PET. Huang reported that the chain folding of nucleation in PTT was ca. 4.8 kcal/mol, which is much lower than that of PET (ca. 10 kcal/mol) [9, 10]. The crystallization rate of poly(trimethylene terephthalate) is faster than that of poly(ethylene terephthalate) [11]. The crystallization behaviors of PET and PTT in the melt and glassy states have been compared [12–14]. Lu and Hay [12] studied the isothermal melt-crystallization and cold-crystallization kinetics of PET with different crystallization temperatures. The Avrami exponents were 2.6 ± 0.2 for both melt-crystallization and cold-crystallization. These results indicated that the crystallization mechanism for both melt-crystallization and cold-crystallization of PET is one of a spherulite being formed by heterogeneous nucleation. Hong et al. [13] investigated the crystallization kinetics and morphology of PTT from the melt and glassy states. The Avrami exponent analyzed from the melt-crystallization process was in the range of 2 ∼ 3, indicating mixed growth and nucleation mechanisms. Chung et al. [14] found that the crystal thickness of cold crystallization (∼2 nm) is about half that of melt crystallization (∼4 nm). The crystal thickness of ∼2 nm is close to the c-axis of the required unit cell for PTT (18.64 Å). They considered that the bundle-like or fringed-micelle crystal was more reasonable than the chain-folded lamella for explaining the crystallite structure of cold-crystallized PTT.
The sequence distribution and crystallization in the melt state of poly(ethylene/trimethylene terephthlate) (ET) copolyesters were investigated [15, 16]. The copolyesters can crystallize across the full range of compositions, and the main constitutional repeating unit in the molecular chain dominates the crystal conformation as a host crystal. The conformational change and crystal structure of ET copolyesters under isothermal melt-crystallization conditions were also investigated [17]. The wide angle X-ray diffraction (WAXD) and in-situ Fourier transform infrared (FTIR) results showed that highly flexible propanediol-dimethyl terephthalate-propanediol (PDO-DMT-PDO) segments in ET53 (EG/PDO: 52.68/47.32) dominated the crystalline structure, a finding that was confirmed by transmission electron microscopy (TEM) observation. However, the crystallization behavior and microstructure of ET copolyesters is not yet understood. The present study investigated the crystallization behavior and morphological structure of ET copolyesters by differential scanning calorimetry (DSC), WAXD, and small-angle X-ray scattering (SAXS) under isothermal melt-crystallization and cold-crystallization conditions.
Experimental
Materials
Poly(ethylene/trimethylene terephthalate) (ET) copolyesters were synthesized with different molar volume ratios of ethylene glycol (EG), 1,3-propanediol (PDO), and dimethyl terephthalate (DMT), using a two-stage reaction sequence. The compositions of the ET copolyesters used are listed in Table 1. The viscosities of ET copolyesters were in the range of 0.7–0.75 dL/g. The details were reported elsewhere [15].
Characterization
For the isothermal crystallization behavior study, the ET copolyesters were melted using a Linkam THMS 600 heater equipped with a LINKAM TMS91 electric microscope controller. The crystallization and melting behaviors were observed with a Du-Pont DSC Q10 instrument using a TA2000 thermal analysis system. The instrument was calibrated with In and Pb. The weight of the samples was in the range of 4–6 mg. The flow rate of the purge gas N2 was approximately 50 cm3/min. Due to the thermal gravimetric analysis of ET copolyesters, there was no significant weight loss of the sample until heating to 300 °C. It was supposed that the transesterification reaction did not happen below 300 °C. Samples were melted at 265 °C, held for 3 min, then rapidly cooled to the isothermal crystallization temperature for 4 hours at 100 °C/min for melt-crystallization. The melt samples were rapidly cooled to 35 °C at 100 °C/min, and then reheated to the isothermal crystallization temperature for 4 hours at 40 °C/min for cold-crystallization. The isothermal crystallization temperatures were set according to the DSC results. Here, ET53 and ET66 were melted at 250 °C to compare them with the isothermal melt-crystallization results in the previous study [17].
The crystal structure of the samples was studied using a Bruker D8 SSS wide-angle X-ray diffractometer with Cu Kα radiation. The electric voltage was 40 kV; electric current was 300 mA; scanning step was 0.02° for 0.5 s; the 2θ scanning angle was 3–70°. The reflection profiles were deconvoluted using Jade 5.0 software, Materials Data, Inc., with a peak search method. The FWHM (full width at half maximum) value used to separate the amorphous phase and crystal reflections was 4.
For the morphological structure study, the small angle X-ray scattering results were obtained using a Bruker NanoSTAR SAXS instrument, which consisted of a Kristalloflex K760 1.5 kW X-ray generator (operated at 40 kV and 35 mA). The cross-coupled Göbel mirrors for Cu Kα-radiation (λ = 1.54 Å) resulted in a parallel beam of about 0.05 mm2 in cross section at the sample position. A Siemens multiwire type area detector with 1024 x 1024 resolution mode was used. All data were corrected by empty beam scattering and the sensitivity of each pixel of the area detector. The area scattering pattern was circularly averaged to increase the efficiency of the data collection. The intensity profile was outputted as the plot of the scattering intensity I(q) vs. the scattering vector, q = 4π/λsin(θ/2) (θ = scattering angle).
Results and discussion
Isothermal crystallization and melting behaviors of ET copolyesters
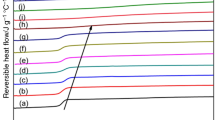
In order to compare the difference in crystallization behavior of ET copolyesters between isothermal melt-crystallization and cold-crystallization, the samples were melt-crystallized and cold-crystallized at the same isothermal temperature (Tc). The copolyesters ET43, ET53, ET66, and ET82 were isothermal at 135, 110, 135, and 150 °C, respectively. After isothermal crystallization for 1 hour, the samples were directly heated at 10 °C/min to melt in order to observe their melting behavior. Figure 1 shows the isothermal melt-crystallization and cold-crystallization and the melting behavior of the ET copolyesters. For the isothermal melt-crystallization, ET82 shows a weak and broad exothermic peak (see Fig. d1). The heat flow increased slightly, although there was no significant exothermic peak in ET43-ET66 (see Figures a1, b1, and c1). When the sample was reheated directly, multiple endothermic peaks in the range of 140-187 °C and 154-240 °C were observed for ET 43 and ET82, respectively (see Figures a2 and d2). A small endotherm for ET53 and ET66 was found in the range of 142-175 °C and 162-188 °C , respectively (see Figures b2 and c2).

Isothermal melt-crystallization and cold-crystallization and melting behaviors of ET copolyesters. Figures a1, b1, c1, and d1 are the DSC curves for isothermal crystallization; Figures a2, b2, c2, and d2 are the reheating DSC curves
The exothermic peaks could be observed clearly under isothermal cold-crystallization conditions for all ET copolyesters (see Fig. 1). The cold-crystallization of the ET copolyesters was nearly completed in less than 30 minutes. The multiple endothermic peaks in the range of 114–188 °C and 140–242 °C were observed in all ET copolyesters when the isothermally crystallized samples were directly reheated. The multiple-melting phenomenon has been widely investigated in the literature [18]. The lowest melting peak temperature (Tm1) is close to the corresponding isothermal crystallization temperature (Tc), in part due to the melting of the secondary crystallites. The low melting peak temperature (Tm2) and the highest melting endothermic temperature (Tm3) are a result of (1) melting of the primary crystallites and the recrystallization and (2) re-melting of the recrystallized crystallites formed during the heating scan, respectively. It can be seen that the endothermic peak (Tm1) for the secondary crystallites existed in the isothermal cold-crystallized ET43, ET53, and ET66 copolyesters. However, the endothermic peak (Tm1) was not observed in the reheating curves of the isothermal melt-crystallized ET53 and ET66 copolyesters. The crystallization exotherm of cold-crystallized copolyester started earlier than that of the corresponding melt-crystallized copolyester, and the enthalpies of crystallization and melting of the cold-crystallized copolyester were larger than those of the corresponding melt-crystallized copolyester. This indicates that the crystallization rate of the isothermal cold-crystallized copolyester was faster than that of the corresponding melt-crystallized copolyester at the same isothermal temperature. This indicates that the nuclei formed during the rapid cooling process at 100 °C/min, and that they enhanced the crystallization behavior of the ET copolyesters during the isothermal cold-crystallization process. Supsphol et al. [18] also reported that the crystallization rate of isothermal cold-crystallized syndiotactic polypropylene (sPP) was faster than that of melt-crystallized sPP at the same isothermal temperature in DSC analysis.
The crystallization kinetics of ET43, ET53, and ET66 under varied isothermal cold-crystallization conditions were investigated using the classic Avrami equation [19–21] as follows:
where X t is the degree of crystallinity at isothermal crystallization time t, n is the Avrami exponent depending on the type of nucleation and growth geometry of the crystals, and k is a rate constant involving both nucleation and growth rate parameters. The kinetic parameters of the ET copolyesters are listed in Table 2. The Avrami exponents (n) of copolyesters are in the range of 2.1 ∼ 2.8, and the crystallization rate constants (k) are plotted at isothermal crystallization temperature in Fig. 2. The crystallization rate constants versus the crystallization temperature of three ET copolyesters are plotted in Fig. 2, and a single bell shape curve can be observed for each copolyester. The maximum crystallization rate constants (k) of ET43, ET53, and ET66 occurred at 130 oC, 120 oC, and 140 oC, respectively. The ideal crystalline structure is interrupted significantly by incorporation of the third constitutional unit into the host crystal and decreases the crystallization rate of ET copolyesters. Supaphol et al. [18] observed double bell shaped curves in melt-crystallized sPP, and described it as the result of nucleation control at a low degree of supercooling and diffusion control at a high degree of supercooling. The cold-crystallization process is dominated by the athermal nucleation mechanism, and therefore these curves exhibit only one maximum for each copolyester. The results of the Avrami exponents and the single bell shaped curves in cold-crystallized ET copolyesters indicate that the cold-crystallization process of ET copolyesters is influenced by the athermal nucleation mechanism. The nuclei formed during the cooling process, which benefits the crystallization of the ET copolyesters during isothermal cold-crystallization.

Crystallization rate constants (k) of ET copolyesters at various isothermal cold-crystallization temperatures
The chain flexibility of PET is lower than that of PTT. The activation energy of ET copolyesters is interesting, and can be calculated approximately using an Arrhenius equation as follows:
where k0 is a temperature-independent pre-exponential factor, ΔEa is the total activation energy, and R is the universal gas constant. The activation energy can be determined from the slope of the Arrhenius plots using (1/n)(ln k) versus 1/T. The activation energies of ET43, ET53, and ET66 were calculated as -11.38, -6.40, and -5.00 kcal/mol, respectively. It is evident that the increase in activation energy of the ET copolyesters is a result of the increase in the EG content.
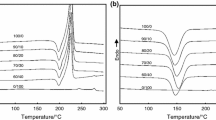
After isothermal cold-crystallization, the sample was reheated directly, showing the multiple melting endothermic peaks of ET copolyester (see Fig. 3). The Hoffman-Weeks extrapolative method was used to estimate the equilibrium melting temperature [22]. Because Tm2 is contributed to the melt of the primary crystallites of copolyesters [18], it was applied to calculate the equilibrium melting temperature using a linear Hoffman-Weeks equation. The equilibrium melting temperatures of the copolyesters obtained from the cold-crystallization of ET43, ET53, and ET66 were 184.6 °C, 172.3 °C, and 229.2 °C, respectively. These results are close to the estimated equilibrium melting temperature of the corresponding ET copolyesters that were determined from the melt-crystallization in our previous work (see Fig. 4) [16]. The equilibrium melting temperature decreased with an increase of the third co-monomer content. A minimum equilibrium melting temperature was obtained in ET53 (174 °C) [16]. We found that although the nucleation mechanism of the melt-crystallization and cold-crystallization processes is different, the equilibrium melting temperatures of both processed copolyesters are similar.

Typical subsequent melting curves of ET43 copolyester after isothermal cold-crystallization at specific temperatures

Hoffman-Weeks plots for melt- and cold-isothermally crystallized ET copolyester. Melt-crystallization data taken from Ref. [16]
Crystal structure of ET copolyesters
The copolyesters were isothermally cold-crystallized for 4 hours (ET82 at 150 °C, ET66 at 105 °C, ET53 at 110 °C, and ET43 at 135 °C), which were selected from our previous study [17], to study their crystal structure. The deconvoluted reflections of the WAXD profiles of these ET copolyesters are shown in Fig. 5. The two broad curves describe the amorphous phase, and all crystal reflections are clearly separated. There are eight PET-related reflections in ET82, and three PET-related reflections in ET66. In ET53, there are six reflections, including two PET-related reflections, three PTT-related reflections, and one new 2θ diffraction peak at ca. 20.67°. In ET43, there are six reflections visible, containing four PTT-related reflections, one PET-related reflection, and a new 2θ diffraction peak at ca. 20.93°. The crystal structures of the ET copolyesters obtained from isothermal cold-crystallization were compared with these obtained from isothermal melt-crystallization at the same isothermal crystallization temperatures [17]. We found that the reflections of ET82 and ET43 obtained from the isothermal cold-crystallization are the same as those from the melt-crystallization. However, one additional PET-related reflection was observed at 11.68° in the cold-crystallized ET53. The highest diffraction intensity in the cold-crystallized ET53 was at 16.48°, which is a PET-related reflection. However the highest diffraction intensity in the melt-crystallized ET53 was at 22.02°, which is a PTT-related reflection. For ET66, two more PET-related reflections were found in cold-crystallized ET66 at 11.92° and 32.07°. The maximum diffraction intensity of cold-crystallized ET53 and ET66 are PET-related reflections, which correspond to the reflection plane of (\( 0\overline{1}1 \)). The new 2θ diffraction peaks at ca. 20.67o in ET53 and 20.93o in ET43 were supposed that the comonomer units included in the crystal lattice to distort the subcell resulting from the inclination of molecular chain.

WAXD deconvolution curves of the ET copolyesters. Samples were isothermally cold-crystallized for 4 hours
The bulk crystallinity was calculated from the deconvoluted reflections of the WAXD profiles using Jade 5 software. The bulk crystallinity of ET82 and ET43 under isothermal cold-crystallization was 33.4 % and 31.9 %, respectively, which is similar to the results from the isothermal melt-crystallized ET82 and ET43 (32.9 % and 31.4 %) [17]. It is obvious that the bulk crystallinity of ET66 and ET53 under isothermal cold-crystallization (10.5 % and 20.3 %) is higher than that under isothermal melt-crystallization, which is 1.9 % and 10.8 %, respectively.
Morphological structure of ET copolyesters
The morphological structures at the lamellar length scale were studied by SAXS. The Lorentz-corrected SAXS profiles of PET (ET100), PTT(ET0), and ET copolyesters after isothermal crystallization for 4 hours are shown in Fig. 6. Here, the PET and PTT were isothermally crystallized at 200 °C and 210 °C, respectively. Other ET copolyesters were isothermally crystallized at the same temperatures as those for the WAXD samples. All melt-crystallized and cold-crystallized samples were found to exhibit a scattering peak except for ET66, which showed the scattering peak only after cold crystallization (see Fig. 6).

Lorentz-corrected intensity profiles of ET copolyesters after isothermal crystallization for 4 hours
The observed scattering peak corresponds to the long period (L) representing the sum of the thickness of the crystalline (lc) and amorphous (la) layers in the lamellar stack, i.e., L = lc + la. The one-dimensional correlation function analysis was adopted to determine lc and la. The one-dimensional correlation function is defined as the Fourier cosine transform of the scattering intensity, viz [23]:
where γ(0) is the scattering invariant.
Since the experimentally accessible scattering vector (q) range is finite, extrapolation of intensities to both low and high q are necessary for the integrations. Extrapolation to the high q values was performed according to the Porod–Ruland model [24–26]:
where K p is the Porod constant, σ is a parameter related to the thickness of the interphase between the crystal and amorphous layers, and I fl is the background intensity arising from the thermal density fluctuation. The values of K p, σ, and I fl are obtained by curve fitting the intensity profile at high q region. Extrapolation to zero is accomplished by the Debye–Bueche model [27]:
where A is a constant and a c is the correlation length. The values of A and a c can be obtained from the plot of I(q)-1/2 versus q2 using the intensity at the low q region.
The one-dimensional correlation function of PET, PTT, and ET copolyesters under isothermal melt-crystallization and cold-crystallization conditions is shown in Fig. 7. It is evident that γ(x) of the cold-crystallized samples generally exhibited a more rapid damping than did the melt-crystallized copolymers, indicating a broader distribution of lamellar thickness. As shown in Fig. 1, the endothermic peak (Tm1) is associated with the melting of the secondary crystallites in the cold-crystallized ET43, ET53, and ET66 copolyesters, and thus the insertion of the secondary crystallites in the lamellar stacks may therefore broaden the lamellar thickness distribution of the cold-crystallized ET copolyesters. Hong et al. also reported that the long period structure of cold crystallized PTT exhibits a shorter range order compared with that of melt-crystallized PTT [14].

The one dimensional correlation function of PET, PTT, and ET copolyesters after isothermal crystallization for 4 hours
The long period L of the lamellar stacks can be determined from the position of the first maximum in the correlation function. The thickness of the thinner layer (l 1 ) is obtained from the intersection between the straight line extended from the self-correlation triangle and the baseline of the minimum of γ(x). The thickness of the thicker layer (l 2 ) is given by l 2 = L-l 1 .
The WAXD results revealed that the bulk crystallinity (i.e., the crystallinity of the sample as a whole) of all copolyesters was lower than 0.5, and thus it is reasonable to assign l 1 = l c and l 2 = l a . When knowing the values of l c and L, the linear crystallinity (or the crystallinity within the lamellar stack) ϕ c lin of the ET copolyesters can be calculated as
Figure 8 plots ϕ c lin as a function of the ET content of the copolymer subjected to melt and cold crystallization. The crystalline lamellar thickness was found to decrease slightly with the increase of the third comonomer content irrespective of the crystallization condition, when the maximum reduction (occurs at 53 % ET content) was ca. 0.4 nm. The linear crystallinity appeared to be independent of the comonomer content, as its value remained at approximately 0.35. For the crystallization of random copolymers, there are two models that describe the location of the comonomer units in the crystallites, the exclusion model and the inclusion model [28]. The exclusion model asserts that the comonomers are excluded from the crystalline lattice. In this case, we expect the crystallite thickness to decrease significantly when the comonomer content increases. In the inclusion model, the comonomers are considered to be included in the crystal lattice, acting as a kind of defect. In that case, the lamellar thickness, although it may be disturbed by the presence of comonomer, may not exhibit as strong a reduction as prescribed by the exclusion model. Our results show that both the crystallite thickness and linear crystallinity depend weakly on the comonomer composition, which strongly implies that the comonomer units in the polymer chains are included in the interior of the crystallite and may serve as the point defects in the lattice.

The long period (L), lamella thickness (l c = l 1 ), amorphous layer thickness (l a = l 2 ), and linear crystallinity (ϕ c lin) of ET copolyesters under isothermal melt-crystallization (filled symbols) and cold-crystallization (open symbols)
Conclusions
The crystal structure, crystallization behaviors, and morphological structure of isothermal melt-crystallized and cold-crystallized ET copolyesters were analyzed in this study. Crystallization of polymer starts at a supercooling. Prior to the isothermal cold-crystallization of the ET copolyester, the sample was rapidly cooled to 35 °C at 100 °C/min, resulting in a non-uniform inner structure of the ET copolyester. The behaviors of isothermal melt-crystallization and cold-crystallization rates of ET copolyesters are different. DSC analyses showed that exothermic peaks can be observed for all of the ET copolyesters under isothermal cold-crystallization condition, while this is not the case under the isothermal melt-crystallization condition. The enthalpies of crystallization and melting of the cold-crystallized copolyester are larger than those of the corresponding melt-crystallized copolyester. The crystallization rate of the ET copolyester at the isothermal cold-crystallization is faster than that of the corresponding ET copolyester at the same isothermal melt crystallization temperature. The Avrami exponents of cold-crystallized ET copolyesters are in the range of 2.1 ∼ 2.8. Single bell shape curves were observed when the crystallization rate constants were plotted with the crystallization temperatures. WAXD analyses showed that the deconvoluted reflections and bulk crystallinity of isothermal cold-crystallized ET53 and ET66 were obviously higher than those of isothermal melt-crystallized ET53 and ET66. This suggests that the nuclei formed during the rapid cooling process at 100 °C/min, where the nuclei benefited from the crystallization behavior of the ET copolyesters during the isothermal cold-crystallization process. The cold-crystallization process of ET copolyesters is influenced by the athermal nucleation mechanism.
SAXS analyses showed a more rapid damping of the one-dimensional correlation function for cold-crystallized ET copolyesters, due to broader lamellar thickness distribution. It was suggested that the insertion of the secondary crystallites, which were abundant in cold crystallization, broadened the thickness distribution. The fact that the crystallite thickness and linear crystallinity did not vary substantially with the composition of the comonomers implied that the comonomer units were included in the crystal lattice as the point defects during both melt-crystallization and cold-crystallization.
References
Wang ZG, Hsiao BS, Fu BX, Liu L, Yeh F, Sauer BB, Chang H, Schultz JM (2000) Correct determination of crystal lamellar thickness in semicrystalline poly(ethylene terephthalate) by small-angle X-ray scattering. Polymer 41(5):1791–1797
Medellín–Rodriguez FJ, Lopez–Guillen R, Waldo–Mendoza MA (1999) Microkinetics of crystallization and melting behavior of solid-state polymerized poly(ethylene terephthalate) (PET). J Polym Sci B Polym Phys 37(16):1981–1990
Zhang W, Shen D (1998) The Effects of Thermal Histories on Crystallization of Poly(ethylene terephthalate). Polym J 30(4):311–314
Hsiao BS, Wang ZG, Yeh F, Gao Y, Sheth KC (1999) Time-resolved X-ray studies of structure development in poly(butylene terephthalate) during isothermal crystallization. Polymer 40(12):3515–3523
Yoo ES, Im SS (1999) Melting behavior of poly(butylene succinate) during heating scan by DSC. J Polym Sci B Polym Phys 37(13):1357–1366
Stein RS, Misra A (1980) Morphological studies on polybutylene terephthalate. J Polym Sci Polym Phys Ed 18(2):327–342
Huang JM, Ju MY, Chu P, Chang FC (1999) Crystallization and melting behaviors of poly(trimethylene terephthalate). J Polym Res 6(4):259–266
Wunderlich B (2003) Reversible crystallization and the rigid–amorphous phase in semicrystalline macromolecules. Prog Polym Sci 28(3):383–450
Huang JM, Chang FC (2000) Crystallization kinetics of poly(trimethylene terephthalate). J Polym Sci B Polym Phys 38(7):934–941
Van Antwerpen F, Van Krevelen DW (1972) Influence of crystallization temperature, molecular weight, and additives on the crystallization kinetics of poly(ethylene terephthalate). J Polym Sci Polym Phys Ed 10(12):2423–2435
Supaphol P, Dangseeyun N, Srimoaon P, Nithitanakul M (2003) Nonisothermal melt-crystallization kinetics for three linear aromatic polyesters. Thermochim Acta 406(1–2):207–220
Lu XF, Hay JN (2001) Isothermal crystallization kinetics and melting behaviour of poly(ethylene terephthalate). Polymer 42(23):9423–9431
Hong PD, Chung WT, Hsu CF (2002) Crystallization kinetics and morphology of poly(trimethylene terephthalate). Polymer 43(11):3335–3343
Chuang WT, Hong PD, Shih KS (2004) Structural formation and gelation behavior of cold-crystallized poly(trimethylene terephthalate). Polymer 45(25):8583–8592
Shyr TW, Lo CM, Ye SR (2005) Sequence distribution and crystal structure of poly(ethylene/trimethylene terephthalate) copolyesters. Polymer 46(14):5284–5298
Shyr TW, Lo CM, Ye SR, Bian J (2007) Sequence distribution and crystal structure of poly(ethylene/butylene terephthalate) copolyesters compared with poly(ethylene/trimethylene terephthalate) copolyesters. J Polym Sci B Polym Phys 45(4):405–419
Shyr TW, Tung CH, Cheng WS, Jiang SD, Yan SK, Gan ZH (2012) The conformational changes, crystal structure and melting behavior of poly(ethylene/ trimethylene terephthalate) copolyesters. Polym Int 61(5):780–787
Supaphol P, Spruiell JE (2001) Isothermal melt- and cold-crystallization kinetics and subsequent melting behavior in syndiotactic polypropylene: a differential scanning calorimetry study. Polymer 42(2):699–712
Avrami M (1939) Kinetics of Phase Change I General Theory. J Chem Phys 7(12):1103
Avrami M (1940) Kinetics of phase change 3. Granulation, phase change, and microstructure. J Chem Phys 9(2):177
Avrami M (1940) Kinetics of Phase Change Transformation-time relations for random distribution of Nuclei. J Chem Phys 8(2):212
Hoffman JD, Weeks JJ (1962) Melting Process and the Equilibrium Melting Temperature of Polychlorotrifluoroethylene. J Res Natl Bur Stand Sect A 66(1):13–28
Strobl GR, Schneider M (1980) Direct evaluation of the electron density correlation function of partially crystalline polymers. J Polym Sci Polym Phys Ed 18(6):1343–1359
Porod G (1951) Die Röntgenkleinwinkelstreuung von dichtgepackten kolloiden Systemen. Colloid Polym Sci 124(2):83–114
Porod G (1952) Die Röntgenkleinwinkelstreuung von dichtgepackten kolloiden Systemen. Colloid Polym Sci 125(1):51–57
Porod G (1952) Die Röntgenkleinwinkelstreuung von dichtgepackten kolloiden Systemen. II. Teil. Colloid Polym Sci 125(2):108–122
Debye P, Bueche AM (1949) Scattering by an Inhomogeneous Solid. J Appl Phys 20(6):518–525
Sanchez IC, Eby RK (1973) Crystallization of random copolymers. J Res Natl Bur Stand Sect A 77A(3):353–358
Acknowledgments
This work is supported by the National Science Council of Taiwan, R.O.C., under grant number 101-2622-E-035-003-CC2. The authors would like to thank Prof. Hsin-Lung Chen, Department of Chemical Engineering, National Tsing Hua University, for his help on the SAXS analysis and discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shyr, TW., Tung, CH., Cheng, WS. et al. The crystallization rate and morphological structure of poly(ethylene/trimethylene terephthlate) copolyesters under isothermal melt-crystallization and cold-crystallization. J Polym Res 20, 186 (2013). https://doi.org/10.1007/s10965-013-0186-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-013-0186-5