
Abstract
A novel self-promoted curing phthalonitrile monomer was synthesized via substitution reaction of 4-nitrophthalonitrile and 3-aminophenol at the presence of K2CO3 in the dimethylsulfoxide solvent. The phthalonitrile was characterized by Fourier transform infrared spectra, nuclear magnetic resonance, gel permeation chromatography, differential scanning calorimetry, dynamic rheological analysis and thermal gravimetric analysis. The phthalonitrile monomer can be thermally polymerized with self-promoted curing behaviors. The prepolymerization reaction of the phthalonitrile prepolymer was investigated and the phthalonitrile prepolymer exhibited the desirable processing feature. With the curing process of low curing temperature and short curing time, the cured polymers exhibited high glass transition temperatures (241–270 °C) and excellent thermal stabilities with the 5 % weight loss temperature (395–441 °C). The novel phthalonitrile can be a good candidate as matrix for high performance polymeric materials.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phthalonitrile resins, as advanced composite matrix materials, have received increasing attention due to their high glass transition temperatures, good mechanical properties, excellent solvent resistance and superior flame resistant properties [1–3]. These resins have been considered as an ideal material for applications in various fields such as marine, aerospace and electronic uses [4–8]. However, the conventional phthalonitriles have the problem of poor processability resulting from their unexpected properties such as high curing temperature, long curing time and narrow processing window (temperature between the melting point and the polymerization temperature) [9, 10]. Therefore, much effort has been focused on improving their processability.
Generally, the neat curing of these phthalonitrile-based monomers carried out very slowly even at the fairly high temperatures and their curing processes have been studied by thermal analysis techniques [11–14]. Therefore, tremendous interests has been focused on initiating the curing process and shortening the curing time of phthalonitrile resins at the present of various curing additives such as phenols [15], organic amines [16], strong organic amines [17] and strong organic acid/amine salts [18]. Thus, the research efforts were concerned mainly with aromatic amines such as bis[4(4-aminophenoxy)phenyl]sulfone (p-BAPS), 1,3-bis(3-aminophenoxy)benzene (m-APB) and 1,4-bis(4-aminophenoxy)benzene (p-APB) [13, 19]. From these results, the utility of aromatic amines is expected to accelerate the polymerization reaction of phthalonitriles. Unfortunately, the addition of aromatic amine curing additives with their volatility at high temperatures had an influence on thermal stability of phthalonitrile-amine compositions.
In order to improve the processability of phthalonitriles without significantly sacrificing their other desirable properties, development of multifunctional phthalonitrile monomers/oligomers is still needed to pursue for advanced applications. Phthalonitrile-based monomers and oligomers containing a variety of linkages such as aromatic ether [10, 20], sulfone [21], oligomeric aromatic ether-containing units [11, 12, 14], oligomeric biphenyl ethernitrile units [22–24] and benzoxazine units [25, 26] have been designed and synthesized. Then, greatly improved properties of these monomers and oligomers can be achieved by including a low complex melt viscosity and a broad processing window. However, the necessity for curing additives in these phthalonitriles has been still recognized to achieve improved processability. In order to obtain better performance for phthalonitriles with good processability, it is desirable to modify the structure of the phthalonitrile monomers with removing the utilization of curing additives. It is well-known that a simple and effective method is incorporating amino or hydroxyl groups into the reactive phthalonitrile units. Then, lots of amino-functional or hydroxy-functional phthalonitrile polymers have been proved to be cross-linked by self-catalyzed curing reaction without the addition of any other curing additives [23].
In this paper, an aromatic amino group was incorporated into the phthalonitrile backbone with the reactive nitrile groups via nucleophilic substitution reaction and this resulted in formation of an amino-containing phthalnitrile monomer. We presented the synthesis, polymerization and processability of the novel thermosetting phthalonitrile. Furthermore, their chemical structure, thermal and thermal-oxide stabilities were studied.
Experimental
Material
All the reagents were analytical grade and were used without further purification. 4-Nitrophthalonitrile (CP) was obtained from Alpha Chemical (Dezhou) Co. Ltd. 3-Aminophenol (CP) and anhydrous potassium carbonate (K2CO3) were obtained from Tianjin BODI Chemicals Co. Ltd. Dimethyl sulfoxide (DMSO) was purchased from Tianjin Guangfu Fine Chemical Research Institute.
Synthesis of 3-aminophenoxyphthalonitrile (3-APN) monomer
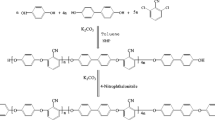
The synthetic route of 3-aminophenoxyphthalonitrile (3-APN) monomer is shown in Scheme 1. The 3-APN monomer was prepared as follow. 4-Nitropathalonitrile (17.30 g, 0.10 mol), 3-aminophenol (10.90 g, 0.10 mol), anhydrous potassium carbonate (17 g, 0.12 mol) and dimethyl sulfoxide (30 ml) were mixed using a stirrer at a stirring speed of 300 rpm. After refluxing at 80 °C for 8 h in nitrogen, the reaction mixture was cooled to room temperature and slowly poured into distilled water in the formation of a solid. The products were filtered and washed several times with distilled water. Then the yellow solid was dried at 70 °C over night after recrystallization from acetone/water (4:1 in volume) mixed solvents (yield: 21.30 g, 91 %). m.p.: 175 °C (DSC).

Synthesis of the 3-APN monomer
1H NMR (300 MHz, DMSO-d 6, δ): 8.06–8.08 (d, 1 H, Ar-H), 7.71 (s, 1 H, Ar-H), 7.33–7.36 (d, 1 H, Ar-H), 7.07–7.12 (t, 1 H, Ar-H), 6.47–6.50 (d, 2 H, Ar-H), 6.23–6.30 (t, 1 H, Ar-H), 5.40 (s, 2 H, NH2).
FTIR (KBr, cm−1): 3445 and 3364 cm−1 (-NH2), 2231 cm−1 (-CN), 1251 cm−1 (stretch, C-O-C), 1490 cm−1 (1, 2 and 4 substitution of benzene ring), 850 cm−1 (1, 3 substitution of benzene ring).
Synthesis of 3-APN prepolymer
The 3-APN prepolymer was prepared in air by heating the 3-APN monomer melt at 180 °C. After 30 min, the reaction was quenched by rapidly cooling the melt to room temperature and the prepolymer was pulverized to use.
1H NMR (300 MHz, DMSO-d 6, δ): 8.05–8.08 (d, 1 H, Ar-H), 7.71–7.70 (d, 1 H, Ar-H), 7.33–7.36 (d, 1 H, Ar-H), 7.07–7.13 (t, 1 H, Ar-H), 6.47–6.50 (d, 2 H, Ar-H), 6.23–6.30 (t, 1 H, Ar-H), 5.41 (s, 2 H, NH2).
FTIR (KBr, cm−1): 3447 and 3364 cm−1 (-NH2), 2232 cm−1 (-CN), 1248 cm−1 (stretch, C-O-C), 1486 cm−1 (1, 2 and 4 substitution of benzene ring), 848 cm−1 (1, 3 substitution of benzene ring).
Synthesis of 3-APN polymer
The same viscous prepolymer was heated and degassed for another 10–20 min until gelation occurred. The gelled samples were poured into these pre-heated molds with cavity dimensions 65 mm × 15 mm × 5 mm, and thermally cured by heating in an oven at 200 °C for 3 h, 200 °C for 6 h, 240 °C for 3 h and 240 °C for 6 h, under ambient conditions, respectively. Then, the 3-APN polymers were obtained.
Measurements
1H NMR spectra
1H NMR spectra were taken on a Bruker AV400 nuclear magnetic resonance spectrometer (NMR) with a proton frequency of 300 MHz and the solvent was DMSO-d 6.
FTIR spectra
FTIR spectra were recorded with Shimadzu FTIR8400S Fourier Transform Infrared spectrometer in KBr pellets between 4000 and 400 cm−1 in air.
Gel permeation chromatography (GPC)
Gel permeation chromatography (GPC) was employed to measure the molecular weights and molecular weight distribution of the 3-APN prepolymer by a PL-GPC220 system using polystyrene as standard and THF as the eluent at the rate of 1.0 ml/min.
Differential scanning calorimetric (DSC)
Differential scanning calorimetric (DSC) analysis was performed by TA Instruments Modulated DSC-Q100 at a heating rate of 10 °C/min and a nitrogen flow rate of 50 ml/min.
Dynamic rheological analysis
Dynamic rheological analysis was performed using TA Instruments Rheometer AR-G2 with a frequency of 1 Hz at different temperatures in air. The samples (0.5–1 g) were melted between 25 mm diameter parallel plates with an environmental testing chamber of the rheometer.
Thermal gravimetric analysis (TGA)
Thermal gravimetric analysis (TGA) was performed on a TA Instruments TGA Q50 with a heating rate of 20 °C/min under nitrogen or air with a purge of 40 ml/min.
Results and discussion
Synthesis and characterization of 3-APN monomer and prepolymer
The 3-APN monomer has been synthesized via substitution reaction of 4-nitrophthalonitrile and 3-aminophenol at the presence of K2CO3 in the dimethylsulfoxide solvent (Scheme 1). Its molecular structures were characterized by 1 H NMR and FTIR spectroscopes. 1 H NMR spectra were shown in Fig. 1 and FTIR spectra were depicted in Fig. 2. The data were found to be in good agreement with the proposed structures. As for the 3-APN monomer, the proton attached to the primary amine (NH2) was observed at 5.04 ppm. Meanwhile, the absorption band at 2231 and 3445 cm−1 are assigned to the stretching vibration of cyano groups (C ≡ N) and amino groups (NH2), respectively. Then assignments of each proton were given to agree well with the proposed molecular structure and all characteristic absorption bands for the expected chemical structures presented in the spectra.

1H NMR spectra of a the 3-APN monomer and b the 3-APN prepolymer

FTIR spectra of the 3-APN monomer and prepolymer
In order to investigate the prepolymerization of the 3-APN monomer, the 3-APN prepolymer was monitored by GPC. The molecular weight of the 3-APN prepolymer was found to be 1000 using polystyrene as the standard (Table 1). The molecular weight was much lower than the values of other thermoplastic reins. However, as a novel thermosetting resin, the prepolymerization of the 3-APN monomer and the preparation of the prepolymer are important to the curing process and processability. In addition, network formation of the 3-APN prepolymer was monitored by 1 H NMR spectroscopes (Fig. 1). Only one additional signal at 7.70 ppm was observed, which may be assigned to triazine unit. Furthermore, the structure of the 3-APN prepolymer also was monitored by FTIR spectra (Fig. 2). The characteristic absorption of the nitrile band at 2231 cm−1 and the amino group at 3445 and 3364 cm−1 decreased. A weak peak assigned to the triazine absorption at 1520 cm−1 was showed [27]. The prepolymerization reaction was predicted to result in triazine ring formation.
Self-promoted curing properties of 3-APN monomer and prepolymer
The self-promoted curing behaviors of the 3-APN monomer were studied by DSC. In the DSC curves shown in Fig. 3, the 3-APN monomer exhibited a single endothermic peak (175 °C), which was attributed to the characteristic melting transition. Then the melting enthalpy of the 3-APN monomer was 127.6 J/g. The exothermic transition peaked at 239 °C and the enthalpy of cure was 179.3 J/g, which were a result of self-promoted curing reaction of the 3-APN monomer between the amino group and phthalonitrile units. For these phthalonitrile monomers [9–21], the polymerization reaction occurs through the nitrile groups of the phthalonitrile units in the presence of aromatic diamine curing agents and these exothermic transitions of the cure reaction peaked at 250–270 °C. In addition, the maximum processing temperature was up to 425 °C and the heat-treatment time was very long (> 10 h) due to the low reactivity of the phthalonitriles. In comparison, the significant reduction in curing temperature of the 3-APN monomer is caused by high concentration of amino groups resulting in the polymerization reaction of the nitrile groups. Polymerization reaction mechanism was shown in Scheme 2. It is important that the 3-APN monomer has lower melting and curing temperatures. Moreover, the pure 3-APN system avoids the volatility of amines at high temperatures.

DSC curves of the 3-APN monomer

Polymerization reaction mechanism of the 3-APN monomer
When the melt sample was cooled down and heated again, the glass transition was observed at 259 °C (second scan) and no edothermic and exothermic peaks were apparent due to melting and curing appeared. For comparison, cooling down and repeated heating revealed the glass transition at 264 °C (third scan) again. These results revealed that the sample of the 3-APN monomer heated during the experiments exhibited relatively high glass transition temperatures and thermal polymerization reaction of the nitrile groups progressed at an extremely slow rate [23]. This is related to the fact that the nitrile curing reaction in the phthalonitrile-amine compositions was controlled as a function of the concentration of the amino or hydroxyl groups containing active hydrogen [15, 21, 28].
The complex melt viscosity changes accompanying the self-promoted curing reaction were monitored by viscosity studies. The time sweep viscosity curves of the 3-APN prepolymer at different temperatures (150 °C, 180 °C and 200 °C) are depicted in Fig. 4. As can be seen, the viscosity of the 3-APN prepolymer was low before the curing reaction occurred and then abruptly increased after the curing reaction started [19]. Moreover, the higher the curing temperature, the faster the viscosity increase. These results indicated that high temperature has a great tendency to accelerate the polymerization reaction of the 3-APN prepolymer. Then, the observed viscosity changes are evidence that the 3-APN prepolymer is reacting to form crosslink networks. As noted in Fig. 5, the storage modulus (G′) and the loss modulus (G″) of the 3-APN prepolymer increased as time increased and the G′ and G″ crossover point corresponding to the gelation time (gel point [29]) was observed at 7 min. These data revealed that the 3-APN prepolymer had been transferred from viscosity flow state to solid state [23]. Meanwhile, the gel point corresponds to the start of cross-linking network formation, which is in consistent with the results got in viscosity studies that the viscosity of the 3-APN prepolymer increased abruptly as gelation occurred. Besides, the tan (delta) curve exhibited a sharp peak at 5 min, which suggested that the 3-APN prepolymer can polymerized without any other curing additives under certain curing temperature. From these rheological data, the 3-APN prepolymer exhibited the desirable processability, which was important to its application in resin transfer molding and resin infusion processes.

Time sweep viscosity curves of the 3-APN prepolymer at different temperatures

Time sweep curves of the 3-APN prepolymer at 180 °C
Thermal properties of 3-APN polymers
Thermally induced phase transition behavior of the 3-APN polymers was investigated by DSC under a nitrogen atmosphere, and their glass transition temperature (T g) are shown in Fig. 6. The 3-APN polymers have high T gs in the range of 241–270 °C, which were attributed to stress relaxation of the polymer network [14]. Obviously, inspection of the T g shows that increasing the curing temperature results in a considerable increase in T g. Furthermore, the T g s of the 3-APN polymers are increased with increasing curing time. Besides, for the DSC curves of the polymers (cured at 200 °C for 3 h, cured at 200 °C for 6 h and cured at 240 °C for 3 h), the exothermic transitions were observed at 259, 262 and 289 °C, respectively, and the corresponding enthalpy decreased from 5.3 J/g, 3.8 J/g and 0.7 J/g. The exothermic transitions correspond to the characteristic triazine ring formation resulting from the cyclotrimerization of the amino-containing phthalonitrile via its nitrile groups [4, 27]. While the exothermic heat flows decreased, it suggested that the reactivity of cyclotrimerization decreased with lower concentration of the reactive groups. Therefore, the high curing temperature and long curing time were necessary for complete conversion to the thermosetting polymers with the high crosslink density.

DSC curves of the 3-APN polymers
FTIR spectroscopy was used to monitor the polymerization reaction of the nitrile groups in the 3-APN polymers (Fig. 7). The FTIR data revealed that the characteristic absorption peak of the nitrile band at 2231 cm−1 gradually decreased, which are attributed to an increase of curing temperature and time. It was consistent with a typical feature of polymerization of phthalonitrile polymers [30]. In addition, a weak peak assigned to the triazine absorption at 1520 cm−1 showed a small increase in intensity as the curing temperature and time increased. Similarly, the observation of the increase in phthalocyanine formation absorbance at 1010 cm−1 was as a function of curing temperature and time, which indicated that the high curing temperature and long curing time were necessary for complete conversion to the thermosetting polymers with the highly aromatic character and heterocyclic ring structures. Then, FTIR studies offer some insight into the self-promoted polymerization reaction and multiple reaction mechanisms of the novel phthalonitrile.

FTIR spectra of the 3-APN polymers
The thermal and thermo-oxidative stabilities of the 3-APN polymers that had been thermally cured without addition of any other curing additives were determined by TGA analysis, and that their curves were shown in Figs. 8 and 9. The 5 % weight loss temperature (T 5%) was measured and the percentage of residue remaining (char yield %) after heating the samples to 800 °C in nitrogen and air atmosphere were listed in Table 2. The T 5% weight losses of the cured 3-APN polymers in nitrogen atmosphere were in the range of 404–430 °C, and char yields at 800 °C were in the range of 71–73 %. In air atmosphere, T 5%s were in the range of 395–441 °C and char yields were in the range of 0.9–17.6 %. According to these results, the thermal and thermo-oxidative stabilities of the cured 3-APN polymers could be increased by the higher curing temperature and longer time, which are necessary for the formation of highly crosslinked networks. However, only for the 3-APN polymer cured at 240 °C for 6 h, the T 5% in air atmosphere is somewhat higher than that in nitrogen atmosphere, which may be ascribed to heterogeneity of the crosslink density. It is related to the different degradation mechanisms and thermal properties according to the thermal history [15, 16]. Therefore, the curing and thermal exposure results showed that the thermal and thermo-oxidative stabilities are a function of the curing temperature and time used in polymerization reaction and the processability of the 3-APN polymers (Table 2).

TGA curves of the 3-APN polymers in nitrogen.

TGA curves of the 3-APN polymers in air
Conclusion
A novel self-promoted curing phthalonitrile was successfully synthesized via substitution reaction. The phthalonitrile monomer can be thermally polymerized at low temperature with its reactive amino groups and nitrile groups and exhibited self-promoted curing behaviors. The phthalonitrile prepolymer exhibited the prepolymerization reaction with the desirable processing feature and the value polydipersity is 1.01. The T g and T 5% of the polymers as function of the curing temperature and time were evaluated to study the self-promoted curing process. These cured polymers exhibited high glass transition temperatures (241–270 °C) and excellent thermal and thermal-oxide stabilities. It is believed that the novel phthalonitrile polymer can be used as a matrix of advanced composites.
References
Shaw SJ (1982) Mater Sci Technol 3:589–99
Hergenrother PM (2003) High Perform Polym 15:3–45
Ho TH, Hwang HJ, Shieh JY, Chung MC (2009) React Funct Polym 69:176–182
Keller TM (1988) J Polym Sci Part A Polym Chem 26:3199–3212
Zhang WX, Wang YZ, Sun CF (2007) J Polym Res 14:467–474
Yang XL, Zhan YQ, Yang J, Zhong JC, Zhao R, Liu XB (2012) J Polym Res 19:9806
Hsiao SH, Huang PC (1997) J Polym Res 4:183–190
Yu GP, Liu C, Li B, Wang LW, Wang JY, Jian XG (2012) J Polym Res 19:9829
Laskoski M, Dominguez DD, Keller TM (2005) J Polym Sci Part A Polym Chem 43:4136–4137
Dominguez DD, Keller TM (2006) High Perform Polym 18:283–304
Laskoski M, Keller TM, Qadri SB (2007) Polymer 48:7484–7489
Laskoski M, Dominguez DD, Keller TM (2007) Polymer 48:6234–6240
Keller TM, Dominguez DD (2005) Polymer 46:4614–4618
Dominguez DD, Keller TM (2007) Polymer 48:91–97
Sumner MJ, Sankarapandian M, McGrath JE, Riffle JS, Sorathia U (2002) Polymer 43:5069–5076
Keller TM, Price TR (1982) J Macromol Sci Chem 18:931–937
Sastri SB, Keller TM (1999) J Polym Sci Part A Polym Chem 37:2105–2111
Burchill PJ (1994) J Polym Sci Part A Polym Chem 32:1–8
Dominguez DD, Keller TM (2008) J Appl Polym Sci 110:2504–2515
Sastri SB, Keller TM (1998) J Polym Sci Part A Polym Chem 36:1885–1890
Keller TM (1994) Chem Mater 6:302–305
Yang XL, Lei YJ, Zhong JC, Zhao R, Liu XB (2011) J Appl Polym Sci 119:882–887
Zeng K, Hong HB, Zhou SH, Wu DM, Miao PK, Huang ZF, Yang G (2009) Polymer 50:5002–5006
Du RH, Li WT, Liu XB (2009) Polym Degrad Stab 94:2178–2183
Zuo F, Liu XB (2010) J Appl Polym Sci 117:1470–1475
Cao GP, Chen WJ, Wei WT, Liu XB (2007) Exp Polym Lett 8:512–518
Sastri SB, Armistead JP, Keller TM (1996) Polym Compos 17:817–822
Moore JA, Han DW (2009) Polymer 50:2551–2557
Laza JM, Vilas JL, Mijangos F, Rodriguez M, Leon LM (2005) J Appl Polym Sci 98:818–824
Jia K, Xu MZ, Zhao R, Liu XB (2011) Polym Int 60:414–421
Acknowledgments
The authors are grateful to the Major Science and Technology Project in Sichuan Province (2010 FZ 0117), “863” National Major Program of High Technology (2012AA03A212) and National Natural Science Foundation (No. 51173021) for the financial supports of this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Guo, H., Chen, Z., Zhang, J. et al. Self-promoted curing phthalonitrile with high glass transition temperature for advanced composites. J Polym Res 19, 9918 (2012). https://doi.org/10.1007/s10965-012-9918-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-012-9918-1