
Abstract
Exfoliated polystyrene/organo-modified montmorillonite nanocomposite was synthesized through in situ free radical polymerization by dispersing a modified reactive organophilic montmorillonite layered silicate in styrene monomers. The original montmorillonite (MMT) was modified by a novel cationic surfactant. A cationic initiator, consisting of a quaternary ammonium salt moiety, α-phenyl chloro acetyl chloride moiety, and 9-decen-1-ol moiety, was intercalated into the interlayer spacing of the layered silicate. Modified MMT clays were then dispersed in styrene monomers and subsequently polymerized by a free-radical in situ polymerization reaction to yield polystyrene/montmorillonite nanocomposite. The structure of obtained modifier was investigated by proton nuclear magnetic resonance (1H NMR) and Fourier-transform infrared (FT-IR) spectroscopy. The exfoliating structure of nanocomposite was probed by X-ray diffraction (XRD) and transmission electron microscopy (TEM). Comparing with pure polystyrene, the nanocomposite showed much higher decomposition temperature and higher glass transition temperature (Tg).
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nanocomposites, composed of clay and polymer, have been studied extensively for some time and it has been shown that most of the properties are enhanced in the presence of a small amount of clay. For instance, incorporation of a few percent of clay in many cases increases the modulus, strength, gas barrier properties, and heat stability, compared to the pure polymers [1–5]. These improvements in the properties are the result of the nanometer scale dispersion of clay in the polymer matrix [6–8]. To make the clay compatible with the polymers, the sodium ions in clay (such as sodium montmorillonite) are usually ion-exchanged with an organic ammonium or phosphonium salt to convert this material into a hydrophobic ammonium or phosphonium treated clay. Although complete compatibility between the long chain of organic modifier and the polymer matrix may be preferable for better dispersion of clay, it appears that the modification of clay by introducing surfactants to obtain better compatibility is less important than the modification of polymer matrix by introducing polar groups. The role of the surfactant is mainly to enlarge the interlayer spacing so to make room for the polymer to penetrate into the gallery space during the preparation of polymer/clay nanocomposites [9, 10].
The preparation of a nanocomposite may be accomplished either by an in-situ polymerization or by blending, with melt blending being the preferred industrial process [11–14]. Nanocomposites are described according to the dispersion of the clay in the polymer. In order to form a nanocomposite, the clay must be uniformly distributed. If this uniform distribution is not achieved, the material is best described as a microcomposite or as an immiscible nanocomposite. In an immiscible nanocomposite the clay is not well-dispersed and it is acting as conventional filler and not as a nano-dimensional material. When the clay is well-dispersed, two different types of nanocomposites may be obtained. If the registry between the clay layers is maintained, intercalated nanocomposites are obtained. If this registry between the clay layers is lost, the nanocomposite is described as delaminated, also known as exfoliated. It is generally accepted that delamination is required for enhanced permeability and mechanical properties while the type of the nanocomposite, intercalated or immiscible, does not seem to be important for thermal properties and fire retardancy of polymer materials [15, 16].
The aims of the present study were the synthesis of polystyrene/montmorillonite nanocomposite via in situ free radical polymerization by using a novel cationic surfactant. This surfactant was synthesized by coupling of α-phenyl chloro acetyl chloride and 9-decen-1-ol in diethyl ether and in the presence of pyridine as a catalyst. Then the product obtained was coupled with trimethylamine in ethanol as the solvent to form the surfactant. Organophilic montmorillonite (O-MMT) was obtained by the ion-exchange process by treating montmorillonite with the surfactant. Finally, modified MMT clays were dispersed in styrene monomers and subsequently polymerized by a free-radical in situ polymerization reaction to yield polystyrene clay nanocomposite.
Experimental
Materials
Styrene (Merck) was distilled under reduced pressure from CaH2 before use. Toluene (Merck) was dried by refluxing over sodium and distilled under argon prior to use. Azobisisobutyronitrile (AIBN), α-phenyl chloro acetyl chloride and 9-decen-1-ol (Merck) were used as received without further purification. Sodium montmorillonite (MMT) was obtained from Southern Clay Products, under the trade name of Cloisite NaC. MMT is reported to have an approximate aspect ratio of 250:1, and is a 2:1 tetrahedral/octahedral aluminum silicate smectite mineral with an idealized chemical formula of Na0.33[Mg0.33Al11.67 Si4O10](OH)2 and a cation exchange capacity of 95 meq/100 g. All other reagents were purchased from Merck and purified according to the standard methods.
Synthesis of undecen-10-enyl-2-chloro-2-phenyl acetate
A solution of α-phenyl chloro acetyl chloride (50 mmol) and diethyl ether (20 ml) was added dropwise, over 1 h, into a mixture of 9-decen-1-ol (50 mmol), pyridine (70 mmol), and diethyl ether (120 ml), and was stirred for 5 h at room temperature. The pyridine–HCl salt that formed was filtered out and washed with a total of 50 ml of diethyl ether. The reaction mixture volume was reduced and washed three times with 30 ml of distilled water. The ether layer was dried over MgSO4 or CaCl2 and then removed to yield clear, light yellow oil. The ester was further purified with silica column chromatography with hexane solvent (Scheme 1).

Synthesis of undecen-10-enyl-2-chloro-2-phenyl acetate
Synthesis of N, N, N-trimethyl-2-oxo-1-phenyl-2-(undecen-10-enyloxy) ethane ammonium chloride
A solution of undecen-10-enyl-2-chloro-2-phenyl acetate (20 mmol) and trimethylamine (30 mmol) in ethanol (50 ml) was stirred at 50°C for 60 h. The ethanol was removed, and the addition of cold ether precipitated the product. The precipitate was washed four times with cold ether (50 ml of each), allowed to sit overnight at -4°C, and then filtered and dried in vacuum oven at room temperature for 48 h (Scheme 2).

Synthesis of N, N, N-trimethyl-2-oxo-1-phenyl-2-(undecen-10-enyloxy) ethane ammonium chloride
Surface modification of montmorillonite
Organophilic montmorillonite was obtained by the ion-exchange process by treating montmorillonite with the synthesized modifier. Montmorillonite was first dispersed in deionized water under ultrasound for 30 min. The synthesized modifier was prepared in deionized water separately, and was added to the clay dispersion at an amount a little higher than the cation exchange capacity (CEC) of montmorillonite. The resulting suspension was intensively stirred for 10 h, and was then filtered and washed with deionized water three times. The final product was dried in vacuum oven at room temperature for 48 h and grounded into powder (Scheme 3).

Surface modification of montmorillonite
Preparation of polystyrene/montmorillonite nanocomposite by in situ radical polymerization
The organophilic montmorillonite was mixed with toluene (10 ml), at a concentration of 3 wt%; and then the mixture was dispersed under ultrasound for 30 min. The initiator (AIBN) and the monomer (styrene) were added to the clay dispersion. The mixture was heated to 80°C in an oil bath and stirred for 24 h, under flowing N2 to complete the polymerization. The mixture was poured into 300 ml methanol for rapid precipitation. The precipitate was filtered and dried at 50°C under vacuum for 2 days. For preparation of pure polystyrene the polymer was cleaved from clay through the refluxing, for about 5 h, of the nanocomposite in a tetrahydrofuran/methanol solution (80/20 v/v) of LiBr (ca. 5 wt%), followed by centrifugation and filtration (Scheme 4).

Preparation of polystyrene/montmorillonite nanocomposite
Characterization
1H NMR spectra were obtained at 25°C on FT-NMR (400 MHz) Brucker spectrometer. Sample for 1H NMR spectroscopy was prepared by dissolving about 10 mg of products in 5 ml of deuterated chloroform. FT-IR spectra were obtained on Shimadzu 8101 M FT-IR. XRD spectra were obtained by using a Siemens D 5000, X-ray generator (CuKα radiation with λ = 1.5406 Å) with a 2θ scan range of 2 to 80° at room temperature. Transmission electron microscopy (TEM) was performed on a Philips EM208 microscope and operated at 100 kV. The sample was prepared by dispersing the powder in ethanol. Imaging was enabled by depositing few drops of suspension on a carbon coated 400 mesh Cu grid. The solvent was allowed to evaporate before imaging. The thermal properties of PS and PS/MMT nanocomposite measurement were performed with a TGA-PL (England). About 10 mg of the sample were heated between 25–600°C, at rate of 10°C min-1, under flowing nitrogen. DSC analyses were carried out using a NETZSCH (Germany)—DSC 200 F3 Maia. The sample was first heated to 200°C and kept for 5 min to eliminate the thermal history. Then the sample was then reheated to 200°C at a rate of 10 °C min-1. The entire test was performed under nitrogen purging at a flow rate of 50 ml min-1.
Results and discussion
Synthesis of modifier
The novel reactive modifier was synthesized by coupling of α-phenyl chloro acetyl chloride and 9-decen-1-ol in diethyl ether solvent and in the presence of pyridine as a catalyst. The product obtained was coupled with trimethylamine in ethanol as the solvent to form new modifier. Figure 1 (a, b) shows the FT-IR spectra of undecen-10-enyl-2-chloro-2-phenyl acetate (a) and N, N, N-trimethyl-2-oxo-1-phenyl-2-(undecen-10-enyloxy) ethane ammonium chloride (b). The FT-IR spectra of undecen-10-enyl-2-chloro-2-phenyl acetate exhibits an absorption band at 1,745 cm-1 attributed to carbonyl stretching bond and absorption bands at 2,855–2,928 cm-1 and 3,070 cm-1 is attributed to –CH aliphatic and aromatic stretching bond respectively. The stretching bands around 1,163 cm-1 and 1,221 cm-1 are due to the C-O group in the ester and the band at 1,664 cm-1 attributed to C = C stretching vibration. Figure (b) exhibits additional absorption bands at 1,024 cm-1 attributed to C-N group and the band around 3,408 cm-1 is due to the ammonium salt group.

FT-IR spectra of undecen-10-enyl-2-chloro-2-phenyl acetate a and N, N, N-trimethyl-2-oxo-1-phenyl-2-(undecen-10-enyloxy) ethane ammonium chloride b
Additional evidence on the synthesis of the new modifier was also obtained from 1H NMR data. The 1H NMR spectra of undecen-10-enyl-2-chloro-2-phenyl acetate indicate the chemical shifts at 1.38–1.64 ppm represent the aliphatic protons (d), and the characteristic signal at 5.36 ppm is assigned to the -CHPhCl proton (benzylic proton) in this product. The 1H NMR spectrum of the new modifier is shown in Figure (b). Comparison with the 1H NMR spectrum of undecen-10-enyl-2-chloro-2-phenyl acetate Figure (b) indicates that the methyl groups of ammonium salt appears with characteristic signals at 3.45 ppm and the proton of –CHPhCl appears at 4.95 ppm. Fig. 2.

1H NMR spectra of undecen-10-enyl-2-chloro-2-phenyl acetate a and N, N, N-trimethyl-2-oxo-1-phenyl-2-(undecen-10-enyloxy) ethane ammonium chloride b
Preparation of organophilic montmorillonite
Figure 3 (a, b) shows the FT-IR spectra of the unmodified montmorillonite (a) and modified montmorillonite (b). In the spectra of both the modified and unmodified clays, the intense peak at 1,045 cm-1 and the two bands at 467 and 525 cm-1 were assigned to Si-O bonds’ stretching and Si-O bonds’ bending, respectively [17, 18]. The bands around 1,637 and 3,630 cm-1 are due to the hydroxyl groups in the clay [19]. The other additional bands observed in the spectrum of the modified clays arise from surfactant bonds’ stretching or bending. The FT-IR spectra of modified clay exhibits an absorption band at 1,740 cm-1 attributed to carbonyl stretching bond. In additionally intercalation of modifier instead of sodium ions can be confirmed by the -CH stretching vibration at 2,855 and 2,925 cm-1.

FT-IR spectra of the unmodified montmorillonite a and modified montmorillonite b
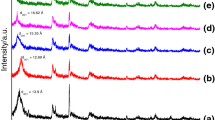
Figure 4 (a, b) shows the XRD patterns of the parent montmorillonite (a) and the organomodified montmorillonite (b). An increase of the basal spacing (d001) of the modified clay is observed after the insertion of the surfactant. More specifically, the pristine montmorillonite shows a d001-spacing of 12.6 Å which correspond to an interlayer space D = 12.6-9.6 = 3 Å, where 9.6 Å is the thickness of the individual clay sheet. In the case of the organomodified montmorillonite, the basal spacing d001 becomes 18.53 Å, with corresponding interlayer space D = 8.93 Å.

XRD patterns of the parent montmorillonite a and the organo-modified montmorillonite b
Preparation of polystyrene/montmorillonite nanocomposite (PS/MMT)
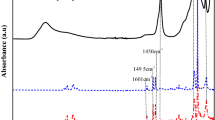
Figure 5 (a, b) shows the FT-IR spectra of the pure polystyrene (a) and polystyrene/montmorillonite nanocomposite (b). The FT-IR spectra of pure polystyrene shows the characteristic absorption bands due to stretching vibration of -CH (3,100-2,850 cm-1), weak aromatic overtone and combination bands in the 2,100-1,650 cm-1 region, C = C stretching vibrations (1,606 cm-1 and 1,510 cm-1), CH2 bending vibrations (1,445 cm-1 and 1,372 cm-1), and γ(C-H) in the aromatic ring (752 cm-1 and 702 cm-1). Apparently the FT-IR spectrum of PS/MMT nanocomposite (Figure b) shows the combination of organomodified montmorillonite and polystyrene. The bands around 1,637 cm-1 and 3,630 cm-1 are due to the hydroxyl groups in the clay. These FT-IR spectra assignments verify that the O-MMT layers have been doped into the polymer matrix during the in situ polymerization and thus form the PS/MMT nanocomposite.

FT-IR spectra of the polystyrene a and polystyrene/montmorillonite nanocomposite b
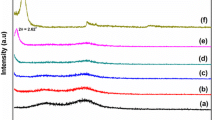
X-ray diffraction, XRD, provides information on the changes of the inter layer spacing of the clay upon the formation of a nanocomposite. The formation of an intercalated structure should result in a decrease in 2θ, indicating an increase in the d-spacing; the formation of an exfoliated structure usually results in the complete loss of registry between the clay layers and no peak can be seen in the XRD trace. In some cases, a disordered immiscible system is obtained and this also shows no peaks, so the absence of an XRD peak cannot be taken as definitive evidence for the formation of an exfoliated nanocomposite and additional evidence, usually transmission electron microscopy (TEM), is required. Figure 6 presents the XRD patterns for the PS/MMT nanocomposite. For the nanocomposite no peaks are observed. Nanocomposite, which could mean either that an exfoliated or an immiscible nanocomposite has been formed.

XRD patterns of PS/MMT nanocomposite
TEM characterization of nanocomposite
Transmission electron microscopy, TEM, provides an actual image of the morphology of the nanocomposite. TEM is complementary to XRD, especially when peaks are not observed in XRD. Figure 7 shows the TEM image for the PS/MMT nanocomposite. The light regions represent polystyrene and the dark lines indicate silicate layers. It is necessary to look at the interactions between clay and polymer, clay and surfactant, and between polymer and surfactant. An optimum balance of all these interactions generally leads to an exfoliated nanocomposite. In the TEM image micronsized clay tactoids are absent because of swelling and expansion by polymer and further exfoliation during polymerization. The TEM image shows that there were mostly well-dispersed silicate layers in the PS matrix. This delineated an exfoliated morphology for the PS/MMT sample.

TEM image for the PS/MMT nanocomposite
Glass transition of PS/MMT nanocomposite
Figure 8 (a, b) shows the DSC traces of the neat PS (a) and PS/MMT nanocomposite (b). The neat PS exhibits an endothermic peak approximately at 75°C, corresponding to the glass transition temperature (Tg). The Tg of the PS/MMT nanocomposite is 79°C, slightly higher than the pure PS. In the addition to that, the glass transition temperature was also increased slightly in the presence of the clay. The increased Tg resulted from the restricted segmental motion of the polymer chain at the organic-inorganic interface, due to the confinement of the PS chains between the silicate layers, as well as the silicate surface polymer interaction in the nano structured hybrids. This result also indicates nano platelets were reasonably well exfoliated and dispersed. It was also reported in the literature that the majority of the other well-dispersed polymer nanocomposites also exhibited higher Tg than their corresponding pristine polymer [20].

DSC traces of the neat PS a and PS/MMT nanocomposite b
Thermal stability of PS/MMT nanocomposite
Characteristic TGA curves of PS and PS/MMT nanocomposite are shown in Figure 9 (a, b). TGA results indicate improvement of the thermal stability for PS/MMT nanocomposite compared to the neat polystyrene. The data of the onset of the degradation temperatures (at which 10% degradation occurs), the midpoint of the degradation temperatures (at which 50% degradation occurs) and the residue that was at 600°C, were shifted to higher in PS/MMT nanocomposite. The residue that was at 600°C, for PS and PS/MMT nanocomposite were 7.26 wt% and 9.83 wt% respectively. Thus 9.83-7.26 = 2.57 wt% remained at 600°C, indicating that the silicate content of this sample was at most 2.57 wt%.

TGA thermograms of PS/MMT nanocomposite a and Pure PS b
The thermal stability as well as the combustion behavior of the polymer-clay nanocomposite is affected by the degradation mechanism of the surfactant molecules. During the thermal treatment of nanocomposite at elevated temperatures, acidic proton sites are formed via the Hoffman decomposition of the alkylammonium modifier of clay, giving protonated montmorillonite that can act as a protonic acid catalyst. Camino et al. ascribed the lower thermal stability of an epoxy resin composite, if compared to the other filled composites, to the larger catalytic activity related to the monoalkyl structure of its organic modifier as compared to the di to tetra alkyl substitution of the other clays [21].
Conclusion
The exfoliated polystyrene/organo-modified montmorillonite nanocomposite was successfully synthesized through in situ free radical polymerization by dispersing montmorillonite that was modified with novel surfactant in styrene monomers. This novel surfactant was synthesized by coupling of α-phenyl chloro acetyl chloride and 9-decen-1-ol and then, the product obtained was coupled with trimethylamine to form the surfactant. The structure of obtained modifier was investigated by 1H NMR and FT-IR spectroscopy.
The XRD and TEM analyses showed that the PS/MMT nanocomposite has a completely exfoliated structure and the O-MMT layers were well dispersed in the polystyrene matrix. The thermal stability and glass transition temperature of PS/MMT nanocomposite improved observably in comparison with pure polystyrene. In conclusion, the unique properties of the nanocomposite result from the strong interactions between the silicate structural layers and the polymeric chains.
Abbreviations
- MMT:
-
Montmorillonite
- O-MMT:
-
Organo-modified montmorillonite
- CEC:
-
Cation exchange capacity
- PS:
-
Polystyrene
- XRD:
-
X-ray diffraction
- AIBN:
-
Azobisisobutyronitrile
- TEM:
-
Transmission electron microscopy
- Tg :
-
Glass transition temperature
- DSC:
-
Differential scanning calorimetry
- TGA:
-
Thermogravimetric analysis
References
Uthirakumar P, Hahn YB, Nahm KS, Lee YS (2005) Eur Polym J 41:1582
Celik M, Onal M (2007) J Polym Res 14(4):313
Chavarria F, Shah RK, Hunter DL, Paul DR (2007) Polym Eng Sci 47:1847
Acharya H, Srivastava SK (2006) Polym Eng Sci 46:837
Sengupta R, Chakraborty S, Bandyopadhyay S, Dasgupta S, Mukhopadhyay R, Auddy K, Deuri AS (2007) Polym Eng Sci 47:1956
Chiu FC, Chu PH (2006) J Polym Res 13(1):73
Zhu J, Wilkie CA (2000) Polym Int 49:1158
Kong Q, Lv R, Zhang S (2008) J Polym Res 15:453
Giannakas A, Spanos CG, Kourkoumelis N, Vaimakis T, Ladavos A (2008) Eur Polym J 44:3915
Jang B, Wilkie CA (2005) Polymer 46:293
Morgan AB, Harris JD (2004) Polymer 45:8695
Samakande A, Hartmann PC, Cloete V, Sanderson RD (2007) Polymer 48:1490
Giannelis E (1996) Adv Mater 8:29
Gilman JW, Jakson CL, Morgan AB, Harris RH, Manias E, Giannelis EP (2000) Chem Mater 12:1866
Sinha RS, Okamoto M (2003) Prog Polym Sci 28:1539
Su S, Jiang DD, Wilkie CA (2004) Polym Degrad Stabil 83:321
Sadhu S, Bhowmick AK (2004) J Appl Polym Sci 92:698
Xu M, Choi YS, Kim YK, Wang KH, Chung IJ (2003) Polymer 44:6387
Choi YS, Ham HT, Chung I (2004) J Chem Mater 16:2522
Li Y, Zhao B, Xie S, Xharng S (2003) Polym Int 52:892
Camino G, Tartaglione G, Frache A, Manferti C, Costa G (2005) Polym Degrad Stabil 90:354
Acknowledgment
The authors express their gratitude to the Bonyade Melli Nokhbeghan institute and Payame Noor University for supporting of this project.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jaymand, M. Surface modification of montmorillonite with novel modifier and preparation of polystyrene/montmorillonite nanocomposite by in situ radical polymerization. J Polym Res 18, 957–963 (2011). https://doi.org/10.1007/s10965-010-9495-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10965-010-9495-0