
Abstract
The nano-SiO2 is used to modify the waterborne polyurethane, and the morphology and performance of the waterborne polyurethane (are studied in) prepared by the in-suit polymerization method and the blending method. The properties and structure have been characterized by fourier transform infrared spectra (IR), differential scanning calorimetry (DSC), Thermal gravimetric (TG), transmittance electron microscopy (TEM) and Dynamical Mechanical Analysis(DMA). The experiment results show that, compared with the blending method, the in-suit polymerization has more advantages in that the nano-SiO2 is evenly dispersed in the waterborne polyurethane, obviously in microphase separation and better in resistance to high temperature and water.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Waterborne polyurethanes (WPU) are high performance polymers known for their excellent properties, such as higher modul,better adhesion, higher heat durability, better elasticity, abrasion resistance, hardness, flexibility, chemical and solvent resistance, gloss, and low temperature film Because a wide range of monomeric materials are now commercially available and tailor-made properties can be obtained from well-designed combinations of monomeric materials, WPU can be tailored to meet the highly diversified demands of modern technologies such as coatings, adhesives, fiber, foams, rubbers, and thermoplastic elastomers, and become the main direction of the modern coating and printing inks development [1].
In recent years, nano structured hybrid organic–inorganic composites based on organic polymer and inorganic nano minerals play an important role in modifying the formation [2, 3]. WPU. The WPU/nano composites have attracted great interest of researchers, because they often exhibit remarkable improvement in materials properties when compared with pure WPU. These improvements include high modul, higher mechanical strength, increased tensile strength and heat resistance, the decreased elongation at break and gas permeability [4–6]. On the other important hand, water dispersions, latexes or emulsions of polyurethane elastomers or coatings permit the application of polyurethanes/nano composites from an aqueous medium. WPU are nontoxic, nonflammable, and do not pollute the air, thus rendering these systems safe with regard to the environment.
The nano-composite material of waterborne polyurethane mainly derives from intercalation polymerization of organo clay or polyurethane/montmorillonite with polyurethane [7, 8], or compounding of carbon nanotube with waterborne polyurethane [9]. Bonding may occur between the hydroxyls in different statuses that exist in SiO2 molecules and the radicals of isocyante and isocyante to improve the performance of polyurethane, e.g. improvement of coating hardness, ductility, compactness, antifriction, high temperature resistance and corrosion resistance. In addition, the network structure of nano-SiO2 formed during coating drying plays a remarkable role in improving the water resistance, aging resistance and finish of coating [10]. Further more, the dispersion modification process of SiO2 nano-material is not complicated, and the raw materials are extensively available at low costs. Thus the study on and production of SiO2 nano-composite material will be set to play a key role in coatings, adhesives and printing ink.
The basic sol–gel process involves the hydrolysis and poly-condensation reactions of metal or silicon alkoxide, or Si(OMe). Metal alkox-ides are hydrolyzed, and metal hydroxides, M(OH) are formed [11, 12]. However, the sol–gel directly incorporated in the waterborne polyurethane, it would lead to production of precipitation.
In this paper, the sol–gel was modified by addition of dihydroxylsilical oil, Silane coupling agents to obtain a nanosilica powder. Whereafter, the composite material of the waterborne polyurethane and the nano SiO2 were prepared by in-suit polymerization of modified silica sol with isocyante and polyester diol, and also via direct dispersion of silica sol into the performed polymer of polyurethane, i.e. blending, and characterization is made using IR, DSC, TG and TEM to study the morphology and performance of the waterborne polyurethane in both in-suit polymerization and blending.
Experimental
Materials
Silica sol (30.5wt.% SiO2 content) with a size of 20nm was supplied by Si Hai Chemical Co.Ltd.(Hubei PR China). Isophorone diisocyanate(IPDI), polycarbonate diol(PCD-1,000), Polycaprolactone (PCL-2,000) and dihydroxy polydimethyl- siloxane(DHPDMS) was purchased from Chuangxin Chemical Co. Ltd (Hubei PR China). Dimethylol propionic acid (DMPA), glycerol, triethylamine (TEA), ethyl acetate(EAC), N-methyl-2-pyrrolodone (NMP), methyl ethylketone (MEK), dibutyltin dilaureate(DBTDL) and isophoronediamina(IPDA) were offered by Sinopharm Chemical Reagent Co. Ltd.
Dispersion and modification of silica sol
Sol–gel was modified as reported [13]. The part of water of sol–gel was removed, then ethanol, dihydroxylsilical oil, and Silane coupling agents were added. The system was disposed and washed repeatly. The modified silica particles were obtained by drying the separated solids.
Preparation of WPU/nanosilica composites
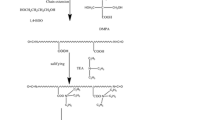
In-situ polymerization method: IPDI and polyols were dried under vacuum at 100°C before use. A 250ml four-necked round-bottomed flask equipped with a mechanical stirrer, a thermometer, a reflux condenser and an inlet of dry nitrogen was used as reactor. Reaction was carried out at a nitrogen atmosphere. Firstly, the nanosilica powder were mixed with NMP (15g) and dispersed by speedy string in the reactor. Then, a stoichiometric mixture of IPDI (14g), PCD-1,000 (13g), PCL- 2,000 (6g), DMPA (3.3g), EAC (10g), 0.05% DBTDL were dropped into the reactor at 70–80°C for 1h. The percent of nanosilica to resin by weight was controlled at contents of 0, 0.5, 1.0, 1.5, 1.8 2.0, 2.5%, respectively. The reaction was carried out at 70–80°C for 1h followed by adding glycerol (0.4g). Afterwards, the system temperature was maintained for 4h until the free isocyanate (NCO) content reached the desired value. The free isocyanate content was determined by the conventional dibutylamine back-titration method [14]. The NCO-terminated prepolymer was obtained by methanol terminating process for 0.5h. Furthermore, TEA was added and stirred for 30min to neutralize the system. The IPDA/H2O solution was added and carefully controlled by a syringe pump under the condition of high speed cutting. Finally, an aqueous dispersion with a solid content of 35wt.% was obtained upon removal of EAC by a rotary evaporator under reduced pressure.
Blending method
The nano silicon powder was dispersed into 5g acetone, and then add deionized water to ultrasonically disperse for 30min under room temperature. Then add this dispersed solution, together with chain extender, into the performed polymer of polyurethane. After high speed cutting and emulsification, a type of waterborne polyurethane nano-composite material is produced.
Characterization of WPU/nanosilica composites
The FT-IR spectra of samples and nanosilica powder were recorded with a Perkin-Elmer Spectrum-2,000 spectrometer in the range of 4,000–400cm−1.
The thermal behaviors of composites was investigated. The differential scanning calorimeter (DSC) analysis was carried out in a liquid nitrogen atmosphere with Perkin-Elmer Pyris 1 DSC analyser. The samples were placed in a vacuum oven at 100°C for 24h, sealed in an aluminum sample cell and held at 100°C for 10min. Then, the samples were quickly put into liquid nitrogen until the temperature reached at −50°C to obtain amorphous samples. These amorphous samples were heated further to 250°C at a heating rate of 10°C/min.
Thermal gravimetric (TG) experiments were performed by simultaneous thermal analyzer (NETZSCH-STA 449C, Germany). Samples were placed in a vacuum oven at 100°C for 24h. And then, 9–12mg samples were placed in an aluminium pan and heated from 20°C to 800°C at a heating rate of 10°C/min. Glass transition temperatures (Tg) were determined from the second heating scan as the mid point of the heat capacity change (δCp).
The morphology of nanosilica particles dispersed in WPU with different particle size and nanosilica content was observed by transmittance electron microscope (HITACHI-JEM-100CXII, Japan). Phosphotungstic acid is used as stainer. Particle size of silica was also measured by TEM (HITACHI-JEM-100CXII, Japan).
Rotating viscosity were measured in a rotating viscosimeter NDJ-1. Measurements were carried out at 25 ± 0.5°C. Each sample was tested for three times, and the values were averaged.
Shear strength tests at composites breaking properties were carried out at a constant crosshead speed set at 50mm/min according to GB/T528-92 standard with an XLL-100A Tensile machine (Taiwan). The shear strength was calculated.
The degree of water absorption was measured by preserving a composites film in water for 24h. The degree of water absorption (%) was calculated by the following equation:
Where W0 represent the original film weight and W represent the film weight after water absorption
Results and discussion
FTIR analysis of WPUN
The FTIR spectrum of WPUNS is shown in Fig. 1. As for WPU/nanosilica composites prepared by blending method, the asymmetric stretching peak of Si–O bonding located at 1,110cm−1 was feeblish, and the C=O stretching peak of urea and urethane located around 1,730–1,600cm−1 was stronger. As for WPU/nanosilica composites by in-suit polymerization method, the stronger Si–O peak located at 1,120cm−1 and C=O peak located around 1,730–1,600cm−1 were observed. As for modified silica particles, a broad characteristic absorbance of –OH group appeared at 3,600–3,400cm−1, and the Si–O peak located at about 1,120–1,020cm−1 was also observed.

FTIR spectrum of a WPUNS by blending method, b WPUNS by in suit polymerization and c modified silica particles
Morphology and particle size distribution of WPUN
The morphology of nanosilica particles dispersed in composites was observed by TEM as shown in Fig. 2 (When we prepare the samples for TEM, the samples have to be diluted to different concentration. The sample (a),(b),(c) are respectively diluted to 1% and sample (d),(e),(f) are diluted to 2%). Generally, as in-situ polymerization method, silica particles can be homogeneously dispersed into polyurethane regardless of the particle size, while some small aggregation of silica particles occurs at relatively high silica concentrations [Figs. 2a,b and c]. The dark spot in Fig. 2a is a result of the vapor bubble during the sample preparing of TEM, when heated under vacuum. On the other hand, as in blending method, only the silica particles with relatively low contents can be evenly dispersed into WPU [Fig. 2d], and the more obvious aggregation was observed as the silica concentration increased [Fig. 2e and f].

TEM of WPUN by in situ polymerization method at nanosilica contents of a 1.0%, b 1.5%, and c 2.5%, respectively, and TEM of WPUN by blending method at nanosilica contents of d 1.0%, e 1.5%, and f 2.5%, respectively
Obviously, compared with WPU/nanosilica composites by in-situ polymerization method, very little amount of silica particles were chemically bonded to WPU matrix in WPUN by blending method [15]. Then, the free nanosilica particles were easy to arouse the aggregation because the residual silanols on silica particle surface increased the probability of further particle growth. This should be the main reason for the above phenomena observed by TEM.
The size distribution of nanosilica particles in WPU emulsion by in-situ polymerization method is shown in Fig. 3. The diameter ranged from 13.2nm to 48.0nm, indicating the homogeneous dispersion of nanosilica particles in WPU emulsion.

The size distribution of nanosilica particles in WPU emulsion by in situ polymerization method
Thermal degradation behaviors and DSC analysis of WPUnanosilica composites
Figure 4 presented the thermal degradation behaviors of WPU/nanosilica composites. It can be seen that the thermal degradation temperatures at maximum weight loss rate ranged from 304.6 to 445.1°C for in-situ polymerization method and from 272.4 to 420.4°C for in blending method. The results suggested that the WPU/nanosilica composites by in-situ polymerization method are more thermally stable than that by blending method. It can be ascribed to the fact that the covalent bonding of silicaol to polyurethane chain accelerated the homogenous dispersion of silica particles and the formation of network structure of nanocomposites, which enhanced the thermal stability.

The thermal degradation behaviors of WPUN
DSC was used to investigate the glass transition temperature (Tg) of soft and hard segments of the composites (Fig. 5). It can be seen that, as the composites by in-situ polymerization, the Tg of soft segment was −30.37°C–45.11°C, and Tg of hard segment was 166.23°C–199.71°C. While, for the composites by blending method, the Tg of soft and hard segments were 28.24°C–48.57°C and 119.16°C–132.11°C, respectively. It is obvious that the wider Tg gap between soft and hard segment was observed for WPUNS by in-situ polymerization method compared with that by blending method. The results indicated that the microphase separation degree of WPUNS by in-situ polymerization was higher than that of WPUNS by blending method because the increased cross-linking degree by the covalent bonding of nanosilica to polyurethane matrix embarrassed the movement of soft and hard segments to some extent.

DSC analysis of WPUN by a in situ polymerization and b blending method (2.0 wt.% silica content)
Static mechanical and physical properties of WPU/nanosilica composites
The static mechanical and physical properties of the composites by blending method are listed in Table 1, and that by in-situ polymerization method listed in Table 2. The data suggested that the shear strength increased and elongation at break decreased with increasing nanosilica content (0.5–2.5%) for both WPU/nanosilica composites. Additionally, it is worth to note that the shear strength of WPU by in-situ polymerization method is higher than that by blending method. As for WPUN by in-situ polymerization method, the higher cross-linking degree by covalent bonding between silica and polyurethane matrix and the further formation of network structure of WPUN attributed to the above result [16]. However, the strength values began to decrease when nanosilica content is over 2.0% for both WPU/nanosilica composites. The reason is mainly attributed to the agglomeration of nanosilica particles in PU.
On the other hand, as for the composites by blending method, the viscosity increased with increasing nanosilica content. One possible explanation is that nanosilica has the higher specific area benefiting the interaction between nanosilica and polyurethane, and the enhanced interaction by higher nanosilica content will result in the increasing viscosity. The same change of viscosity was also observed for the composites by in-situ polymerization method. The increased cross-linking degree with higher nanosilica content mentioned above should be the main reason for this result.
Furthermore, the enhanced resistance to water was observed with increasing nanosilica content for the composites by both methods. It seems due to that the higher nanosilica content will increase the amounts of free paths of water molecules to pass through WPU/nanocomposites matrix and reduce water absorption [17].
Conclusions
A waterborne polyurethane nano-composite material is produced via in-suit polymerization and blending separately. In in-suit polymerization, the IR spectrum shows a strong absorption peak of Si–O bond while a weak one in blending. The morphological analysis indicates that the in-suit polymerization method deliver better dispersion of nano-SiO2 in waterborne polyurethane than blending, and also provides better resistance to high temperature and water of the composite material of the waterborne polyurethane and the nano-SiO2.
Abbreviations
- DBTDL:
-
dibutyltin dilaureate
- DSC:
-
Differential Scanning Calorimetry
- DHPDMS:
-
dihydroxy polydimethylsiloxane
- DMPA:
-
dimethylol-propionic acid
- DMA:
-
Dynamical Mechanical Analysis
- EAC:
-
ethyl acetate
- FTIR:
-
Fourier Transform Infrared Spectra
- IPDI:
-
isophorone diisocyanate
- MEK:
-
Methyl ethylketone
- NMP:
-
N-methyl-pyrolidone
- PCL:
-
polycapro- lactone
- PCD:
-
polycarbonate diol
- WPU:
-
polyurethane
- TEA:
-
triethylamine
- TGA:
-
Thermogravimetric Analysis
- TEM:
-
Transmittance Electron Microscopy
- IPDA:
-
isophorone diamine
References
Harjunalanen T, Lahtinen M (2003) The effects of altered reaction conditions on the properties of anionic poly(urethane-urea) dispersions and films cast from the dispersions. Eur Polym J 39:817–824. doi:10.1016/S0014-3057(02)00279-3
Huybrechts J, Bruylants P, Vaes A, De A (2000) Surfactant-free emulsions for waterborne, two-component polyurethane coatings. Prog Org Coat 38:67–77 doi:10.1016/S0300-9440(00)00083-7
Wicks ZW, Wicks DA, Rosthauser JW (2002) Two package waterborne urethane systems. Prog Org Coat 44:161–183. doi:10.1016/S0300-9440(02)00002-4
Hou MH, Liu WQ, Li Y, Chen JH (2005) Waterborne polyurethane doubly modified by montmorillonite and siloxane. Yingyong HuaxueChin 22:1132–1136
Hou MH, Liu WQ, Li Y, Chen JH (2005) Preparation and characterization of waterborne polyurethane/silane montmorillonite nanocomposite. Shiyou HuagongChin 34:677–680
Kuan HC, Ma CC, Chang WP, Yuen SM, Wu HH, Lee TM (2005) Synthesis, thermal, mechanical andrheological properties of multiwall carbon nanotubewaterborne polyurethane nanocomposite. Compos Sci Technol 65:1703–1710. doi:10.1016/j.compscitech.2005.02.017
Kuan HC, Chuang WP, Ma CC (2005) Synthesis and characterization of a clay/waterborne polyurethane nanocomposite. J Mater Sci 40:179–185. doi:10.1007/s10853-005-5704-3
Jeong HM, Jang KH, Cho K (2003) Properties of waterborne polyurethanes based on polycarbonate diol reinforced with organophilic clay, journal of macromolecular science. Physics B 42:1249–1263
Kwon JY, Kim HD (2005) Preparation and properties of acid-treated multiwalled carbon nanotube/waterborne polyurethane nanocomposites. J Appl Polym Sci 96:595–604. doi:10.1002/app.21436
Mamunya YeP, Shtompel VI, Lebedev EV, Pissis P, Kanapitsas A, Boiteux G (2004) Structure and water sorption of polyurethane nanocomposites based on organic and inorganic components. Eur Polym J 40:2323–2331. doi:10.1016/j.eurpolymj.2004.06.007
Kuan HC, Su HY, Ma CC (2005) Synthesis and characterization of polysilicic acid nanoparticles/waterborne polyurethane nanocomposite. J Mater Sci 40:6063–6070. doi:10.1007/s10853-005-1302-7
Yano S, Hick K, Kurita K (1998) Physical properties and structure of organic–inorganic hybrid materials produced by sol–gel process. Mater Sci Eng C 6:75–90. doi:10.1016/S0928-4931(98)00043-5
Jesionowski T (2002) Krysztaf kiewicz, A, preparation of the hydrophilic/hydrophobic silica particles, colloids surf A. physicochem. Eng Aspects 207:49–58. doi:10.1016/S0927-7757(02)00137-1
Cong SF, Yu LR (2003) Polyurethane coating, firsted, chemistry industrial publisher ltd., Perkin. Chapter 11:288
Chen YC, Zhou S, Yang HH, Gu GX, Wu LM (2004) Preparation and characterization of nanocomposite polyurethane. J Colloid Interface Sci 279:370–378. doi:10.1016/j.jcis.2004.06.074
Zhou SX, Wu LM, Sun J, Shen WD (2002) The change of the properties of acrylic-based polyurethane via addition of nano-silica. Prog Org Coat 45:33–42. doi:10.1016/S0300-9440(02)00085-1
Chen TK, Tien YI, Wei KH (2000) Synthesis and characterization of novel segmented polyurethane/clay nanocomposites. Polymer (Guildf) 41:1345–1353. doi:10.1016/S0032-3861(99)00280-3
Acknowledgements
The authors are grateful for the financial support from the Natural Science Foundation of Hubei Province in China (Grants 2006ABA174).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chen, JJ., Zhu, CF., Deng, HT. et al. Preparation and characterization of the waterborne polyurethane modified with nanosilica. J Polym Res 16, 375–380 (2009). https://doi.org/10.1007/s10965-008-9238-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10965-008-9238-7