
Abstract
In this research, manganese ferrite (MnFe2O4) nanoparticles (NPs) were synthesized using the sol-gel hydrothermal process through different hydrothermal durations and polyvinyl alcohol (PVA) contents. The synthesized NPs were characterized by X-ray diffraction (XRD), vibrating sample magnetometry (VSM), and field emission scanning electron microscopy (FESEM) techniques. The effect of adding PVA as a surfactant and hydrothermal treatment duration on phase formation, microstructure, and magnetic properties of NPs have been discussed in this article. Rietveld-refined XRD patterns proved formation of magnetite and nonstoichiometric manganese ferrite phases along with MnFe2O4 in samples. The existence of a hematite phase was observed in samples prepared without PVA at the hydrothermal durations of 15 and 20 h, while adding PVA caused the hematite phase to disappear. FESEM images showed that adding PVA caused angular and coarser particles to form. The hysteresis loops of the samples confirmed the ferrimagnetic behavior of some samples while others presented superparamagnetic characteristics depending on their processing conditions. The saturation magnetization (Ms) and magnetic coercivity (Hc) values of NPs varied between 38 and 64 emu/g and 15–85 Oe, respectively. Addition of PVA with the average hydrothermal duration of 7.5 h provides the sample with the highest Ms (equal to 64 emu/g), which is mainly composed of MnFe2O4, Mn1.03Fe1.97O4, and Fe3O4. On the other hand, the sample P10 prepared with PVA addition at the hydrothermal treatment duration of 10 h presented the highest value of MnFe2O4 according to the Rietveld refinement results of XRD patterns performed by Maud program.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Manganese ferrite (MnFe2O4) is a soft magnetic material with a reverse spinel structure belonging to the space group of Fd3m. Spinels are magnetic oxides with general formula of AB2O4 where A and B are divalent and trivalent cations, respectively, occupying either octahedral or tetrahedral sites. Oxygen anions occupy FCC sites in this structure. If all divalent cations occupy tetrahedral sites, the structure is defined as a normal spinel structure. When octahedral sites are occupied by a fraction of divalent cations, equivalent amounts of trivalent cations occupy tetrahedral sites. This structure is defined as a partially inverted spinel structure [1]. MnFe2O4 is a partial inverse spinel ferrite in which 80% of Mn ions occupy tetrahedral (A) sites and the remaining 20% of ions occupy octahedral (B) sites [2, 3]. This material has attracted much attention because of its appropriate properties such as high chemical stability, proper mechanical hardness [4], high magnetic permeability, high saturation magnetization (Ms), and low loss [5]. Particle size and crystal morphology play a major role in properties and application of materials that are strongly dependent to synthesis method and its conditions. Magnetic interactions between nanoparticles (NPs) provide novel properties in comparison with bulk material due to their small volume and high surface to volume ratio [6]. MnFe2O4 NPs are used in many areas such as high-density information storage [7], biosensors [8], drug delivery [9], magnetic resonance imaging [10], ferrofluids, gas sensors [11], and many other high-technological applications.
MnFe2O4 is widely used as a sensing material due to its multiple valence states of Mn. Vignesh et al. [12] presented the promising properties of MnFe2O4 NPs for toxic gas, ammonia, sensing application operating at room temperature with high sensitivity and selectivity towards ammonia. MnFe2O4 is also used as a photocatalyst for environmental applications including elimination of azo dyes from wastewater, because of its excellent properties such as great optical absorption over low energy photon, chemical stability, large surface area to volume ratio, and high Ms [13]. In addition, MnFe2O4 NPs are advantageous candidates for magnetic hyperthermia applications and direct tumor targeting due to their high magnetization, high chemical stability, high specific loss power, and low toxicity [14,15,16]. These NPs are also capable of binding to drugs, proteins, enzymes, antibodies, or nucleotides, and can be heated in alternating magnetic fields [17]. Aghajanzadeh et al. [18] have shown that MnFe2O4 NPs are suitable for anticancer drug delivery of therapeutic drugs. Moreover, MnFe2O4 NPs are used as a powerful negative contrast agent for multimode magnetic resonance imaging of cancer cells with the ability to enhance surrounding water proton signals on the T1-weighted image [19].
Several methods have been used to synthesize MnFe2O4 NPs such as chemical bath deposition [20], chemical co-precipitation [21], combustion [22], hydrothermal [23], reverse micelle process [24], thermal decomposition [25], mechanochemical [26], solvothermal [27], high-energy wet milling [28], sol-gel [29], sonochemical [30], and sol-gel hydrothermal [31] techniques. Gas condensation, microemulsion, laser vaporization, and vacuum deposition and vaporization are other techniques used to synthesize nano-crystalline ferrites [32, 33].
All the methods used for synthesis of nano-sized ferrites have their advantages and shortcomings. The inert gas aggregation method is a simple, continuous, and flexible method for producing ferrites and multi-component NPs such as multi-core/shell structures, providing ultrafine-grained materials with narrow grain size distribution, clean grain boundaries, high purity, and excellent resistance to grain growth. However, the method suffers from some drawbacks such as agglomeration of particles, high cost, and difficulty of maintaining a clean vacuum. The method is slow and limited to a laboratory scale. It also requires rather high vapor pressure metals and presents a source-precursor incompatibility, temperature variations, and dissimilar evaporation rates [34, 35]. Likewise, vacuum deposition and vaporization have advantages such as high deposition rates and economical yield; however, many compounds are difficult to be deposited using this technique. Additionally, the laser vaporization technique presents several advantages compared to other heating techniques by producing a high density of vapor in a very short time, which is useful for direct deposition of particles [32, 36, 37]. Nevertheless, laser evaporation is less effective in the evaporation of a metallic target in comparison with an oxide target and the high Ms of the particles leads to high agglomeration tendency. Laser evaporation may also alter the mean particle size and magnetic phase of the sample in the evaporation chamber [38].
Chemical co-precipitation is widely used for the synthesis of magnetic NPs and is a simple, low-cost, environmentally friendly, and high yield method, which provides homogeneous and size-controlled particles of materials [11, 14]. Co-precipitation provides composition flexibility of products and large-scale preparation; nevertheless, the tendency of agglomeration is one of the issues concerning the synthesis of magnetic NPs by this method [39, 40]. Moreover, high pH value of the reaction mixture has to be controlled in both the synthesis and purification steps, which leads to less uniform and monodisperse NPs [41]. Thermal decomposition, another promising way for the preparation of magnetic NPs, provides particles with a very narrow size distribution as well as controlled size and morphology [42]. The issues faced to this method are the necessity for laborious purification steps and surface treatment [17]. The microemulsion process, as well, has many advantages such as ease of preparation, thermodynamic stability, minimal agglomeration of nano-sized particles, narrow size distribution, and composition control. Though, the shortcomings of this method include requiring large amounts of surfactants and limited solubilizing capacity for substances with high melting points [42, 43]. Also, sonochemical synthesis of ferrite particles has nanometric size range, moderate Hc, and low remanence ratio, making them more appropriate for application in electronic gadgets [30]. However, the powders prepared by the sonochemical method are usually porous, amorphous, and agglomerated [17]. Additionally, major drawbacks of the milling procedure are the difficulty of adjusting the desired particle size and shape. The milling procedure also leads to lattice defects that change the magnetic properties of particles [42].
The sol-gel method is a multi-purpose method widely used for synthesis of nanomaterials due to its simplicity, low cost, and high efficiency [44]. Final products prepared by this method are homogeneous and relatively high in purity. The sol-gel method enables synthesis at a low temperature and allows for fine control of the product’s chemical composition. Despite these, the method is relatively time-consuming and some organic solvents may be harmful to the human body [43]. The hydrothermal method has many advantages for synthesis of nanomaterials because of its low synthesis temperature, well-crystallized products, and simplicity of controlling various factors, such as time of treatment, stirring rate, size, morphology, agglomeration, and composition of products [45,46,47].
The sol-gel hydrothermal synthesis provides advantages of both sol-gel and hydrothermal methods. This method has attracted much attention for several advantages provided in the synthesis of nanomaterials, such as high purity, high crystallinity, narrow particle size distribution, low synthesis temperature, excellent compositional control, homogeneity on the molecular level, and controlled morphology of the final products [48,49,50].
Providing monodispersed particles in nanoscales is a considerable issue in the synthesis of nano-ferrites. This issue could be managed by polyvinyl alcohol (PVA) addition, which plays a noteworthy role in the synthesis of nanomaterials by controlling polymerization of the gel, NPs growth, morphology, agglomeration, and producing NPs with uniform shapes because of its unique structure, which forms steric layers against aggregation and particle growth [51,52,53]. PVA as a surfactant also reduces the interfacial tension of the solution or emulsion and provides stabilization of mixture in the first step of particle preparation [51]. Up to now, there has not been any research about the influence of adding PVA and hydrothermal treatment duration on phase formation, microstructure, and magnetic properties of MnFe2O4 NPs synthesized by the sol-gel hydrothermal process. In this paper, synthesis procedure, phase formation, microstructure, and magnetic properties of different samples synthesized with and without PVA addition using different hydrothermal treatment durations have been investigated.
2 Experimental
2.1 Materials
All chemicals used in this study were of analytical grade obtained from Merck Co. and were used as received without further purification. Materials used for synthesis of MnFe2O4 NPs were manganese acetate (Mn(CH3COO)2 .4H2O, 99% Merck) and iron(III) chloride (FeCl3, 98% Merck) as metal sources, citric acid (C6H8O7.H2O, 99.5% Merck) as a homogeneous distributer of metal ion, sodium hydroxide (NaOH, 98% Merck) as a mineralizer, and PVA (98% Merck) as a surfactant.
2.2 Synthesis of MnFe2O4 NPs
To prepare 0.01 mol MnFe2O4, 2.4509 g of manganese acetate powder, 3.2440 g of iron chloride powder, and 2.1014 g of citric acid were dissolved in 25 ml of distilled water separately (the molar ratio of Mn:Fe was 1:2 and the molar ratio of manganese acetate:citric acid was 1:1). After complete dissolution, citric acid solution was poured into the manganese acetate solution to obtain a homogeneous aqueous solution after stirring. Afterwards, 0.1 vol.% PVA was added to some particular samples while stirring. Iron chloride solution was added to the mixture and the mixture was stirred for more 30 min. Finally, the NaOH solution was gradually added to the obtained sol to reach pH to 12. The mixture was stirred for 1 h while condensation reactions turned it to a dark brownish gel. The obtained gel was poured into a Teflon-lined stainless steel autoclave and was placed in an oven for different hydrothermal treatment durations of 5, 7.5, 10, 15, and 20 h at 180 °C. The precipitates were filtered and washed several times with distilled water and were dried for 4 h using an oven at 80 °C. Table 1 illustrates the structure of sample coding based on the absence or presence of PVA and duration of hydrothermal treatment.
2.3 Experimental Techniques and Data Treatment
The phase formation of NPs has been studied by a Philips PW1730 X-ray diffractometer (XRD) using Cu Kα radiation (λ = 0.1541 nm). The XRD patterns were submitted to a quantitative analysis by the Rietveld method using MAUD (material analysis using diffraction) software to obtain weight percentage of phases as well as their lattice parameters.
Magnetic properties of the samples were investigated at room temperature using a vibrating sample magnetometer (VSM) model MDK6 at maximum applied field of 10 kOe. The morphology of the samples was studied using a field emission scanning electron microscope (FESEM) model Mira 3-XMU.
3 Results and Discussion
3.1 XRD Analysis
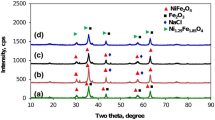
XRD patterns of MnFe2O4 NPs synthesized with and without PVA by the sol-gel hydrothermal technique at 180 °C and hydrothermal durations of 5, 7.5, 10, 15, and 20 h are presented in Fig. 1. To determine the structural features of the samples, Rietveld refinement of XRD patterns has been performed using the MAUD program. Figure 2 shows Rietveld refinement of XRD data of the N5 sample. In Fig. 2, the black spheres represent experimental data; the red solid line shows calculated values obtained by the Rietveld method; and the black line at the lower box shows residuals.

XRD patterns of MnFe2O4 NPs synthesized with and without PVA at 180 °C and hydrothermal durations of 5, 7.5, 10, 15, and 20 h

Rietveld-refined XRD pattern of sample N5. Black spheres are experimental data, and the solid red line represents the Rietveld refinement. The black line in the lower box shows the residuals
Phase analysis associated with the Rietveld refinement of XRD patterns assured formation of a variety of phases with different weight percentages in different samples. Table 2 provides a detailed information of all the phases observed in samples. The mean crystallite size of the three as-mentioned phases was calculated using the Debye-Scherrer equation.
where L is the crystallite size, λ is the X-ray wavelength of Cu Kα radiation = 0.1541 nm, θ is the Bragg diffraction angle of the highest peak, and β is the full width of the highest peak at half maximum (FWHM) in radian after instrumental broadening correction [52]. The intensity of the main diffraction peak of cubic spinel ferrites, MnFe2O4 and Fe2.937O4, at the (311) plane and the intensity of the main diffraction peak of α-Fe2O3 at the (104) plane were considered as a measure of their degree of crystallinity.
The weight percentage for the phases of different samples was calculated with the Rietveld method. Phase content, weight percentage, and mean crystallite size of all samples are listed in Table 3.
XRD patterns of the obtained diffraction patterns ensured formation of MnFe2O4 with a spinel structure in all samples with different quantities. The XRD pattern of the MnFe2O4 coincides well with the standard data of the cubic spinel MnFe2O4 (Jacobsite) phase (ICDD card no. 01-088-1965). The obtained peaks indexed to the (111), (220), (311), (222), (400), (422), (511), (440), and (533) crystal planes of the MnFe2O4. On the other hand, the resultant Rietveld-refined XRD patterns showed that formation of Mn1.03Fe1.97O4, Mn0.43Fe2.57O4, Fe3O4 (magnetite), and Fe2O3 (hematite) also occurs with different weight percentages in different samples. The results of XRD patterns illustrate that by increasing hydrothermal durations of samples prepared without PVA, the quantity of MnFe2O4 phase decreases. At higher hydrothermal treatment durations of 15 h and 20 h of samples synthesized without PVA (N15 and N20), an additional phase named hematite with a rhombohedral structure (ICDD card no. 01-079-1741) appeared and progressively increased in amount with the increase of hydrothermal duration. The corresponding observed peaks of hematite phase could be indexed as (012), (104), (110), (113), (024), (116), (214), and (300) diffraction planes. Increasing hydrothermal duration causes the decomposition of MnFe2O4 phase and formation of hematite phase as it has been seen by M. Jalalian et al., which was reported for cobalt ferrite at high hydrothermal durations [53]. As a result, remained manganese ions must have formed the Mn1.03Fe1.97O4 phase or have been dissolved in the water of the system and have been washed out from products through filtration.
In contrast with the samples prepared without PVA, samples synthesized with PVA present higher MnFe2O4 content with higher hydrothermal durations until the hydrothermal duration of 10 h. These samples do not show a noticeable percentage of hematite phase in their XRD patterns. It seems that in sample P5, the pressure of the system and reaction time were not adequate to perform ion exchanges in crystal structure and form desirable phase structure [54]. Increasing hydrothermal treatment duration in the samples prepared with PVA causes the Mn1.03Fe1.97O4 phase to decrease in content and leads to the formation of higher quantities of MnFe2O4 at sample P10 with more than 90 wt.% at shorter hydrothermal durations than that reported by Ibrahim et al., which was equal to 48 h [31]. Probably, in sample P10, PVA prevents decomposition of MnFe2O4 and formation of additional phases. PVA reduces the surface tension of the solution and lowers the energy needed for the formation of MnFe2O4 [55]. In addition, as hydroxyl groups of PVA can make hydrogen bonds with anions, the solubility of the metal salt in the solution increases which leads to the growth of crystallites. Moreover, the polymeric network of PVA prohibits cation mobility and leads to the maintenance of local stoichiometry as well as reducing the formation of additional undesirable phases [56].
Due to the rather high similarity of the XRD patterns of different spinel structures, overlapping of magnetite and MnFe2O4 diffraction peaks could be possible in the obtained patterns.
The estimated results of Table 3 showed that the mean crystallite size of samples varies between 24 and 46 nm. The lowest crystallite size was observed at the hydrothermal treatment duration of 7.5 h in both samples prepared with and without PVA. Although the variation of crystallite size does not show any sequence in both series of samples, which could be due to the formation of a variety of phases in each sample, the results revealed that increasing hydrothermal treatment time favors the growth of the crystallite size. Increasing reaction time causes pressure of the system to increase and provides more time for metal ions to transfer and form spinel structure. Therefore, with the increased reaction time from 7.5 to 15 h in the samples prepared with and without PVA, the size of crystallites and the system crystallinity increase [54]. In the hydrothermal duration time of 20 h in sample N20, MnFe2O4 crystallites have most likely been decomposed and some of them have been turned to hematite, which made hematite crystallites to disport from MnFe2O4 crystallites and caused the particle size reduction. Crystallite size of samples synthesized with PVA is higher than that of samples prepared without PVA in the same hydrothermal treatment times, which could explain the role of PVA in facilitating crystallite growth. PVA increases the reaction rate and solubility of metal ions and enhances their tendency to form crystal structure or crystallite growth [56]. PVA accelerates the reaction kinetics, forcing nanocrystals to be formed at shorter hydrothermal treatment durations and increases their growth rate and size [53].
3.2 FESEM Investigations
Figure 3 shows the microstructure of samples. The matching between the crystallite sizes calculated with the Debye-Scherrer formula and that obtained from FESEM images is quite reasonable. All of the samples have nano-sized particles. The increase of particle size is observed as the hydrothermal treatment duration increases, which can be due to the increase of the formation of Fe2+ ions accelerating the growth of grain size. These images demonstrate that NPs have tendency to form agglomerates because of the strong magnetic interactions between ferrite particles, high reactivity of particles, and formation of inter-particle London-van der Waals bonds either in the wet or dry state due to their fine particle size [22, 57,58,59]. In addition, it can be seen that samples, which were synthesized with PVA, have more irregular and coarser shapes. PVA molecules can modulate the growth rate along certain directions with high surface energy, which causes a variety of nanocrystal shapes [60]. For instance, a proper amount of CTAB surfactant often induces the synthesis of triangle Fe3O4 nanocrystals [61].

FESEM images of N5 (a), N7.5 (b), N10 (c), N15 (d), N20 (e), P5 (f), P7.5 (g), P10 (h), and P15 (i) nano-powders
Particle formation involves many steps, which can be influenced by the surfactant. The reaction rate, product, and stereochemistry may significantly vary from those observed in dilute aqueous solution [62].
3.3 VSM Studies
Figure 4 presents hysteresis curves of the samples. Values of Ms and magnetic coercivity (Hc) are listed in Table 4.

Magnetization curves of samples
The Ms value of all samples is beneath the Ms value of the bulk MnFe2O4 that is equal to 77 emu/g [63]. The reduction in Ms value is due to the spin canting effect in the disordered surface layers of NPs with large surface to volume ratio [64,65,66]. Ms in spinel ferrites is influenced by many factors such as preparation route, cation distribution, crystallite size, particle size, and sintering conditions calculated by the exchange interactions between octahedral and tetrahedral sites. Total Ms is equal to difference of magnetic moments of these two sites [55, 66]. According to Neel’s model, as occupancy of B sites with Fe3+ or Mn2+ ions increases, the Ms value increases. In contrary, as the occupancy of A sites with magnetic ions increases, the Ms value decreases [67]. Ms values also decreased with the decrease of crystallite size and the increase of spin canting [68, 69].
Mostafa et al. reported the value of 31.08 emu/g for the Ms of MnFe2O4 NPs synthesized by hydrothermal route at 180 °C in the presence of NaOH as a mineralizer [70]. The reported Ms value for nano-sized MnFe2O4 synthesized by mechanosynthesis is about 49.77 emu/g [71]. MnFe2O4 NPs synthesized using sonochemical synthesis have also shown the values of 15, 18, and 22 emu/g for Ms of particles with the size of 34, 42, and 46 nm, respectively, which were prepared in different calcination temperatures [30]. The Ms value of 69 emu/g was also reported for MnFe2O4 NPs prepared via the low-power ultrasonic-assisted co-precipitation method [72]. Zhang et al. have also reported the Ms value of 54.5 emu/g for MnFe2O4 NPs synthesized by a co-precipitation method in the presence of CTAB [73].
Ms values of the samples synthesized without PVA addition range from 38 to 62 emu/g, which does not show any sequence with hydrothermal duration variations; however, Hc values increase with the increase of hydrothermal duration from 15 to 85 Oe. As is shown in Table 4, sample N5 has a large Ms value which may be due to the existence of magnetite phase with a large Ms of 92 emu/ that is much larger than the Ms of MnFe2O4 (77 emu/g) [63]. At the hydrothermal duration of 7.5 h, Ms reduces. This is due to the elimination of magnetite phase and formation of a great deal of Mn1.03Fe1.97O4 with lower Ms than that of magnetite or MnFe2O4. The lower Ms also may be due to the lower crystallite size and effect of spin canting in the disordered surface layers and lower crystallization [74]. At the hydrothermal duration of 10 h, the Ms increases, which could be due to the existence of the MnFe2O4 phase with larger crystallite sizes than N5 and N7.5. Ms has been extensively reduced at the hydrothermal duration of 15 h. This phenomenon is most likely due to the presence of Mn1.03Fe1.97O4 and formation of hematite phase, which is a nonmagnetic phase.
Ms values of samples with PVA addition show an increase until hydrothermal duration of 7.5 h from 53 to 64 emu/g, and then it decreases to 52 emu/g. Ms values of samples P5, P10, and P15 are in the same range and are smaller than those of sample N5, which may be due to the existence of larger amounts of Mn1.03Fe1.97O4 and antiferromagnetic hematite (in sample P10) with lower Ms values [75, 76]. Sample P7.5 shows the highest value of Ms, which is equal to 64 emu/g. This sample presents large values of magnetite phase with higher Ms compared to other manganese iron oxide phases. On the other hand, polymer molecules are probable to form bonds with the surface of ferrite NPs and reduce the surface spin disorder, which leads to the increase in Ms. As a result, higher Ms values of the samples prepared with PVA can be explained [27].
Hc values of the samples prepared with PVA do not show any sequence and are higher than those of bulk sample which is equal to 20 Oe [22]. Hc is affected by surface distortions due to the crystal defects and dislocations. Variation of Hc may be due to the effect of size, disordered spins in the surface layer, ion exchange, and cation distribution variations, which causes network strain [54, 57, 71].
In order to show the status of our results and compare the magnetic properties of our prepared MnFe2O4 NPs with other ferrite NPs reported in a variety of literatures, we have performed a comparative study on the magnetic values and particle size of MnFe2O4 NPs and other ferrites prepared by different synthesis methods in Table 5. Comparing the results of the table below indicates that our MnFe2O4 NPs synthesized by the simple and low-cost sol-gel hydrothermal process present great magnetic properties as well as nano-sized particle size compared with other techniques used to prepare magnetic ferrites. The maximum value of Ms (64 emu/g) obtained from the sample with PVA addition and hydrothermal duration of 10 h is amongst the highest values of Ms achieved from other techniques in literatures. In addition, in terms of Hc evaluation, our synthesized NPs with different processing parameters presented different magnetic behaviors: some presented ferrimagnetic and some presented superparamagnetic characteristics.
4 Conclusion
In this research, MnFe2O4 NPs were synthesized using the sol-gel hydrothermal process at 180 °C. Samples were synthesized either with or without PVA addition with treatment durations of 5, 7.5, 10, 15, and 20 h. Phase content of samples was largely affected by the hydrothermal treatment duration and PVA addition. The Rietveld refinement results of XRD patterns have shown that alongside with the MnFe2O4 phase, a variety of undesirable phases such as nonstoichiometric manganese ferrite and magnetite phases form in the samples. Samples synthesized without PVA addition had a secondary phase (hematite) formed at hydrothermal durations of 15 and 20 h while with PVA addition, this phase eliminated in the XRD patterns. The hysteresis loops of the samples confirmed the ferrimagnetic behavior of some samples while others exhibited superparamagnetic behavior depending on their processing conditions. The Ms and Hc of the samples were between 38 to 64 emu/g and 15 to 85 Oe, respectively. Microstructural studies represented that all the samples were of nanometer grade and PVA addition led to the formation of coarser particles. Microstructure studies illustrated spherical particles for the samples synthesized without PVA addition and angular particles for the samples synthesized with PVA addition. Also calculated mean crystallite sizes of the samples by the Debye-Scherrer equation show an increasing trend from 7.5 to 15 h of hydrothermal treatment duration with the lowest crystallite size attributed to the samples N7.5 and P7.5. Addition of PVA with the average hydrothermal duration of 7.5 h provides the sample with the highest Ms (equal to 64 emu/g), which is mainly composed of MnFe2O4, Mn1.03Fe1.97O4, and Fe3O4. On the other hand, the sample P10 prepared with PVA addition at the hydrothermal treatment duration of 10 h, presented the highest value of MnFe2O4 according to the Rietveld refinement results of XRD patterns performed by MAUD program.
References
Gaudon, M., Pailhé, N., Wattiaux, A., Demourgues, A.: Structural defects in AFe2O4 (A= Zn, Mg) spinels. Mater. Res. Bull. 44(3), 479–484 (2009)
Kadam, R., Biradar, A., Mane, M., Shirsath, S.E.: Sol-gel auto-combustion synthesis of Li3xMnFe2− xO4 and their characterizations. J. Appl. Phys. 112(4), 043902 (2012)
Gorter, E.W.: Saturation magnetization and crystal chemistry of ferrimagnetic oxides. I. II. Theory of ferrimagnetism. Philips Res. Rep. 9(295-320), 321–365 (1954)
Lu, J., Ma, S., Sun, J., Xia, C., Liu, C., Wang, Z., Zhao, X., Gao, F., Gong, Q., Song, B.: Manganese ferrite nanoparticle micellar nanocomposites as MRI contrast agent for liver imaging. Biomaterials. 30(15), 2919–2928 (2009)
Arulmurugan, R., Jeyadevan, B., Vaidyanathan, G., Sendhilnathan, S.: Effect of zinc substitution on Co–Zn and Mn–Zn ferrite nanoparticles prepared by co-precipitation. J. Magn. Magn. Mater. 288, 470–477 (2005)
Hamdeh, H.H., Ho, J., Oliver, S., Willey, R., Oliveri, G., Busca, G.: Magnetic properties of partially-inverted zinc ferrite aerogel powders. J. Appl. Phys. 81(4), 1851–1857 (1997)
Musat, V., Potecasu, O., Belea, R., Alexandru, P.: Magnetic materials from co-precipitated ferrite nanoparticles. Mater. Sci. Eng. B. 167(2), 85–90 (2010)
Haun, J.B., Yoon, T.J., Lee, H., Weissleder, R.: Magnetic nanoparticle biosensors. Wiley interdisciplinary reviews. Nanomed. Nanobiotechnol. 2(3), 291–304 (2010). https://doi.org/10.1002/wnan.84
Kumar, C.S., Mohammad, F.: Magnetic nanomaterials for hyperthermia-based therapy and controlled drug delivery. Adv. Drug Deliv. Rev. 63(9), 789–808 (2011)
Sahoo, B., Devi, K.S.P., Dutta, S., Maiti, T.K., Pramanik, P., Dhara, D.: Biocompatible mesoporous silica-coated superparamagnetic manganese ferrite nanoparticles for targeted drug delivery and MR imaging applications. J. Colloid Interface Sci. 431, 31–41 (2014)
Zipare, K., Dhumal, J., Bandgar, S., Mathe, V., Shahane, G.: Superparamagnetic manganese ferrite nanoparticles: synthesis and magnetic properties. J. Nanosci. Nanoeng. 1(3), 178–182 (2015)
Vignesh, R.H., Sankar, K.V., Amaresh, S., Lee, Y.S., Selvan, R.K.: Synthesis and characterization of MnFe2O4 nanoparticles for impedometric ammonia gas sensor. Sensors Actuators B Chem. 220, 50–58 (2015)
Rahmayeni, R., Oktavia, Y., Stiadi, Y., Arief, S., Zulhadjri, Z.: Spinel ferrite of MnFe2O4 synthesized in Piper betle Linn extract media and its application as photocatalysts and antibacterial. J. Dispers. Sci. Technol. 42, 1–10 (2020)
Makridis, A., Topouridou, K., Tziomaki, M., Sakellari, D., Simeonidis, K., Angelakeris, M., Yavropoulou, M.P., Yovos, J.G., Kalogirou, O.: In vitro application of Mn-ferrite nanoparticles as novel magnetic hyperthermia agents. J. Mater. Chem. B. 2(47), 8390–8398 (2014)
Patade, S.R., Andhare, D.D., Somvanshi, S.B., Jadhav, S.A., Khedkar, M.V., Jadhav, K.: Self-heating evaluation of superparamagnetic MnFe2O4 nanoparticles for magnetic fluid hyperthermia application towards cancer treatment. Ceram. Int. 46(16), 25576–25583 (2020)
Chandunika, R., Vijayaraghavan, R., Sahu, N.K.: Magnetic hyperthermia application of MnFe2O4 nanostructures processed through solvents with the varying boiling point. Mater. Res. Express. 7(6), 064002 (2020)
Majidi, S., Zeinali Sehrig, F., Farkhani, S.M., Soleymani Goloujeh, M., Akbarzadeh, A.: Current methods for synthesis of magnetic nanoparticles. Artif. Cells, Nanomed. Biotechnol. 44(2), 722–734 (2016)
Aghajanzadeh, M., Naderi, E., Zamani, M., Sharafi, A., Naseri, M., Danafar, H.: In vivo and in vitro biocompatibility study of MnFe2O4 and Cr2Fe6O12 as photosensitizer for photodynamic therapy and drug delivery of anti-cancer drugs. Drug Dev. Ind. Pharm. 46(5), 846–851 (2020)
Wang, Z., Liu, J., Li, T., Liu, J., Wang, B.: Controlled synthesis of MnFe 2 O 4 nanoparticles and Gd complex-based nanocomposites as tunable and enhanced T 1/T 2-weighted MRI contrast agents. J. Mater. Chem. B. 2(29), 4748–4753 (2014)
Sharma, U.S., Sharma, R.N., Shah, R.: Physical and magnetic properties of manganese ferrite nanoparticles. Int. J. Eng. Res. Appl. 4(8), 14–17 (2014)
Augustin, M., Balu, T.: Synthesis and characterization of metal (Mn, Zn) ferrite magnetic nanoparticles. Mater. Today: Proc. 2(3), 923–927 (2015)
Deraz, N., Alarifi, A.: Controlled synthesis, physicochemical and magnetic properties of nano-crystalline Mn ferrite system. Int. J. Electrochem. Sci. 7, 5534–5543 (2012)
Phumying, S., Labuayai, S., Swatsitang, E., Amornkitbamrung, V., Maensiri, S.: Nanocrystalline spinel ferrite (MFe2O4, M= Ni, Co, Mn, Mg, Zn) powders prepared by a simple aloe vera plant-extracted solution hydrothermal route. Mater. Res. Bull. 48(6), 2060–2065 (2013)
Hashim, M., Shirsath, S.E., Meena, S., Mane, M., Kumar, S., Bhatt, P., Kumar, R., Prasad, N., Alla, S., Shah, J.: Manganese ferrite prepared using reverse micelle process: structural and magnetic properties characterization. J. Alloys Compd. 642, 70–77 (2015)
Gao, R.-R., Zhang, Y., Yu, W., Xiong, R., Shi, J.: Superparamagnetism and spin-glass like state for the MnFe2O4 nano-particles synthesized by the thermal decomposition method. J. Magn. Magn. Mater. 324(16), 2534–2538 (2012)
Bellusci, M., Aliotta, C., Fiorani, D., La Barbera, A., Padella, F., Peddis, D., Pilloni, M., Secci, D.: Manganese iron oxide superparamagnetic powder by mechanochemical processing. Nanoparticles functionalization and dispersion in a nanofluid. J. Nanopart. Res. 14(6), 1–11 (2012)
Aslibeiki, B., Kameli, P., Ehsani, M., Salamati, H., Muscas, G., Agostinelli, E., Foglietti, V., Casciardi, S., Peddis, D.: Solvothermal synthesis of MnFe2O4 nanoparticles: the role of polymer coating on morphology and magnetic properties. J. Magn. Magn. Mater. 399, 236–244 (2016)
Şimşek, T., Akansel, S., Özcan, Ş., Ceylan, A.: Synthesis of MnFe2O4 nanocrystals by wet-milling under atmospheric conditions. Ceram. Int. 40(6), 7953–7956 (2014)
Hou, X., Feng, J., Ren, Y., Fan, Z., Zhang, M.: Synthesis and adsorption properties of spongelike porous MnFe2O4. Colloids Surf. A Physicochem. Eng. Asp. 363(1-3), 1–7 (2010)
Goswami, P.P., Choudhury, H.A., Chakma, S., Moholkar, V.S.: Sonochemical synthesis and characterization of manganese ferrite nanoparticles. Ind. Eng. Chem. Res. 52(50), 17848–17855 (2013)
Ibrahim, I., Ali, I.O., Salama, T.M., Bahgat, A., Mohamed, M.M.: Synthesis of magnetically recyclable spinel ferrite (MFe2O4, M= Zn, Co, Mn) nanocrystals engineered by sol gel-hydrothermal technology: high catalytic performances for nitroarenes reduction. Appl. Catal. B Environ. 181, 389–402 (2016)
Rajput, N.: Methods of preparation of nanoparticles-a review. Int. J. Adv. Eng. Technol. 7(6), 1806 (2015)
Scano, A., Ennas, G., Frongia, F., La Barbera, A., López-Quintela, M.A., Marongiu, G., Paschina, G., Peddis, D., Pilloni, M., Vázquez-Vázquez, C.: Mn–ferrite nanoparticles via reverse microemulsions: synthesis and characterization. J. Nanopart. Res. 13(7), 3063–3073 (2011)
Suryanarayana, C., Prabhu, B.: Synthesis of nanostructured materials by inert-gas condensation methods. In: Nanostructured Materials, pp. 47–90. Elsevier (2007). https://doi.org/10.1016/B978-081551534-0.50004-X
Grammatikopoulos, P., Steinhauer, S., Vernieres, J., Singh, V., Sowwan, M.: Nanoparticle design by gas-phase synthesis. Adv. Phys.: X. 1(1), 81–100 (2016)
Iles, G.N., Baker, S., Thornton, S., Binns, C.: Enhanced capability in a gas aggregation source for magnetic nanoparticles. J. Appl. Phys. 105(2), 024306 (2009)
Oprea, B., Martínez, L., Román, E., Vanea, E., Simon, S., Huttel, Y.: Dispersion and functionalization of nanoparticles synthesized by gas aggregation source: opening new routes toward the fabrication of nanoparticles for biomedicine. Langmuir. 31(51), 13813–13820 (2015)
Moras, K., Schaarschuch, R., Riehemann, W., Zinoveva, S., Modrow, H., Eberbeck, D.: Production and characterisation of magnetic nanoparticles produced by laser evaporation for ferrofluids. J. Magn. Magn. Mater. 293(1), 119–126 (2005)
Indira, T., Lakshmi, P.: Magnetic nanoparticles–a review. Int. J. Pharmaceut. Sci. Nanotechnol. 3(3), 1035–1042 (2010)
Islam, K., Haque, M., Kumar, A., Hoq, A., Hyder, F., Hoque, S.M.: Manganese ferrite nanoparticles (MnFe2O4): size dependence for hyperthermia and negative/positive contrast enhancement in MRI. Nanomaterials. 10(11), 2297 (2020)
Wu, W., He, Q., Jiang, C.: Magnetic iron oxide nanoparticles: synthesis and surface functionalization strategies. Nanoscale Res. Lett. 3(11), 397–415 (2008)
Biehl, P., Von der Lühe, M., Dutz, S., Schacher, F.H.: Synthesis, characterization, and applications of magnetic nanoparticles featuring polyzwitterionic coatings. Polymers. 10(1), 91 (2018)
Rane, A.V., Kanny, K., Abitha, V., Thomas, S.: Methods for synthesis of nanoparticles and fabrication of nanocomposites. In: Synthesis of inorganic nanomaterials, pp. 121–139. Woodhead Publishing (2018). https://doi.org/10.1016/B978-0-08-101975-7.00005-1
Zhu, Y.-F., Du, R.-G., Chen, W., Qi, H.-Q., Lin, C.-J.: Photocathodic protection properties of three-dimensional titanate nanowire network films prepared by a combined sol–gel and hydrothermal method. Electrochem. Commun. 12(11), 1626–1629 (2010)
Suchanek, W.L., Riman, R.E.: Hydrothermal synthesis of advanced 668 ceramic powders. In: Advances in Science and Technology, vol. 45, pp. 184–193. Trans Tech Publications Ltd (2006). https://doi.org/10.4028/www.scientific.net/AST.45.184
Boulos, M., Guillemet-Fritsch, S., Mathieu, F., Durand, B., Lebey, T., Bley, V.: Hydrothermal synthesis of nanosized BaTiO3 powders and dielectric properties of corresponding ceramics. Solid State Ionics. 176(13-14), 1301–1309 (2005)
Chen, Z., Zhan, G., Wu, Y., He, X., Lu, Z.: Sol–gel-hydrothermal synthesis and conductive properties of Al-doped ZnO nanopowders with controllable morphology. J. Alloys Compd. 587, 692–697 (2014)
Yu, H., Ouyang, S., Yan, S., Li, Z., Yu, T., Zou, Z.: Sol–gel hydrothermal synthesis of visible-light-driven Cr-doped SrTiO 3 for efficient hydrogen production. J. Mater. Chem. 21(30), 11347–11351 (2011)
Singhal, S., Namgyal, T., Singh, J., Chandra, K., Bansal, S.: A comparative study on the magnetic properties of MFe12O19 and MAlFe11O19 (M= Sr, Ba and Pb) hexaferrites with different morphologies. Ceram. Int. 37(6), 1833–1837 (2011)
Brunacci, N., Wischke, C., Naolou, T., Neffe, A.T., Lendlein, A.: Influence of surfactants on depsipeptide submicron particle formation. Eur. J. Pharm. Biopharm. 116, 61–65 (2017)
Rao, J.P., Geckeler, K.E.: Polymer nanoparticles: preparation techniques and size-control parameters. Prog. Polym. Sci. 36(7), 887–913 (2011)
Cullity, B.D., John, W.: Weymouth: elements of X-ray diffraction. Am J Phys. 25(6), 394–395 (1957)
Jalalian, M., Mirkazemi, S., Alamolhoda, S.: The effect of poly vinyl alcohol (PVA) surfactant on phase formation and magnetic properties of hydrothermally synthesized CoFe2O4 nanoparticles. J. Magn. Magn. Mater. 419, 363–367 (2016)
Yao, L., Xi, Y., Xi, G., Feng, Y.: Synthesis of cobalt ferrite with enhanced magnetostriction properties by the sol− gel− hydrothermal route using spent Li-ion battery. J. Alloys Compd. 680, 73–79 (2016)
Yalçın, O., Bayrakdar, H., Özüm, S.: Spin-flop transition, magnetic and microwave absorption properties of α-Fe2O4 spinel type ferrite nanoparticles. J. Magn. Magn. Mater. 343, 157–162 (2013)
Barakat, N.A., Park, S.J., Khil, M.S., Kim, H.Y.: Preparation of MnO nanofibers by novel hydrothermal treatment of manganese acetate/PVA electrospun nanofiber mats. Mater. Sci. Eng. B. 162(3), 205–208 (2009)
Angermann, A., Töpfer, J., Da Silva, K., Becker, K.: Nanocrystalline Mn–Zn ferrites from mixed oxalates: synthesis, stability and magnetic properties. J. Alloys Compd. 508(2), 433–439 (2010)
Hsiang, H.-I., Tsai, J.-Y.: Titanate coupling agent effects on nonaqueous Co 2 Z ferrite suspensions dispersion. J. Mater. Sci. 41(19), 6339–6346 (2006)
Rak Z.S.: From nanosize powders to a diesel soot converter. In: Baraton M.I., Uvarova I. (eds) Functional gradient materials and surface layers prepared by fine particles technology. NATO Science Series (Series II: Mathematics, Physics and Chemistry), vol 16. Springer, Dordrecht (2001). https://doi.org/10.1007/978-94-010-0702-3_9
Jun, Y.W., Choi, J.S., Cheon, J.: Shape control of semiconductor and metal oxide nanocrystals through nonhydrolytic colloidal routes. Angew. Chem. Int. Ed. 45(21), 3414–3439 (2006)
Shi, R., Gao, G., Yi, R., Zhou, K., Qiu, G., Liu, X.: Controlled synthesis and characterization of monodisperse Fe3O4 nanoparticles. Chin. J. Chem. 27(4), 739–744 (2009)
Dixit, S.G., Mahadeshwar, A.R., Haram, S.K.: Some aspects of the role of surfactants in the formation of nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 133(1-2), 69–75 (1998)
Özgür, Ü., Alivov, Y., Morkoç, H.: Microwave ferrites, part 1: fundamental properties. J. Mater. Sci. Mater. Electron. 20(9), 789–834 (2009)
Li, J., Yuan, H., Li, G., Liu, Y., Leng, J.: Cation distribution dependence of magnetic properties of sol–gel prepared MnFe2O4 spinel ferrite nanoparticles. J. Magn. Magn. Mater. 322(21), 3396–3400 (2010)
Mosivand, S., Monzon, L., Kazeminezhad, I., Coey, J.M.D.: Influence of growth conditions on magnetite nanoparticles electro-crystallized in the presence of organic molecules. Int. J. Mol. Sci. 14(5), 10383–10396 (2013)
Mohammed, E., Malini, K., Kurian, P., Anantharaman, M.: Modification of dielectric and mechanical properties of rubber ferrite composites containing manganese zinc ferrite. Mater. Res. Bull. 37(4), 753–768 (2002)
Mostafa, N.Y., Zaki, Z., Heiba, Z.: Structural and magnetic properties of cadmium substituted manganese ferrites prepared by hydrothermal route. J. Magn. Magn. Mater. 329, 71–76 (2013)
Hochepied, J., Pileni, M.: Magnetic properties of mixed cobalt–zinc ferrite nanoparticles. J. Appl. Phys. 87(5), 2472–2478 (2000)
Kumar, L., Kumar, P., Kar, M.: Cation distribution by Rietveld technique and magnetocrystalline anisotropy of Zn substituted nanocrystalline cobalt ferrite. J. Alloys Compd. 551, 72–81 (2013)
Mostafa, N.Y., Hessien, M., Shaltout, A.A.: Hydrothermal synthesis and characterizations of Ti substituted Mn-ferrites. J. Alloys Compd. 529, 29–33 (2012)
Bolarín-Miró, A.M., Vera-Serna, P., Sánchez-De Jesús, F., Cortés-Escobedo, C.A., Martínez-Luevanos, A.: Mechanosynthesis and magnetic characterization of nanocrystalline manganese ferrites. J. Mater. Sci. Mater. Electron. 22(8), 1046–1052 (2011)
Pourbafarani, S., Mozaffari, M., Amighian, J.: Investigation of phase formation and magnetic properties of Mn ferrite nanoparticles prepared via low-power ultrasonic assisted co-precipitation method. J. Supercond. Nov. Magn. 26(3), 675–678 (2013)
Zhang, Y., Nan, Z.: Modified magnetic properties of MnFe2O4 by CTAB with coprecipitation method. Mater. Lett. 149, 22–24 (2015)
Šepelák, V., Bergmann, I., Menzel, D., Feldhoff, A., Heitjans, P., Litterst, F., Becker, K.: Magnetization enhancement in nanosized MgFe2O4 prepared by mechanosynthesis. J. Magn. Magn. Mater. 316(2), e764–e767 (2007)
Chlan, Vaojtěch.: Hyperfine Interactions in Ferrites with spinel structure. Praha, 2013. Rigorózní práce. Univerzita Karlova, Matematicko-fyzikální fakulta, Katedra fyziky kondenzovaných látek. http://hdl.handle.net/20.500.11956/60890. Accessed October 11 2020
Lin, C.-S., Hwang, C.-C., Huang, T.-H., Wang, G.-P., Peng, C.-H.: Fine powders of SrFe12O19 with SrTiO3 additive prepared via a quasi-dry combustion synthesis route. Mater. Sci. Eng. B. 139(1), 24–36 (2007)
Gubbala, S., Nathani, H., Koizol, K., Misra, R.: Magnetic properties of nanocrystalline Ni–Zn, Zn–Mn, and Ni–Mn ferrites synthesized by reverse micelle technique. Phys. B Condens. Matter. 348(1-4), 317–328 (2004)
Victory, M., Pant, R., Phanjoubam, S.: Synthesis and characterization of oleic acid coated Fe–Mn ferrite based ferrofluid. Mater. Chem. Phys. 240, 122210 (2020)
Son, S., Swaminathan, R., McHenry, M.: Structure and magnetic properties of rf thermally plasma synthesized Mn and Mn–Zn ferrite nanoparticles. J. Appl. Phys. 93(10), 7495–7497 (2003)
Kanitz, A., Hoppius, J.S., del Mar Sanz, M., Maicas, M., Ostendorf, A., Gurevich, E.L.: Synthesis of magnetic nanoparticles by ultrashort pulsed laser ablation of iron in different liquids. ChemPhysChem. 18(9), 1155–1164 (2017)
Vijayakumar, R., Koltypin, Y., Felner, I., Gedanken, A.: Sonochemical synthesis and characterization of pure nanometer-sized Fe3O4 particles. Mater. Sci. Eng. A. 286(1), 101–105 (2000)
Yetim, N.K., Baysak, F.K.U., Koç, M.M., Nartop, D.: Characterization of magnetic Fe3O4@SiO2 nanoparticles with fluorescent properties for potential multipurpose imaging and theranostic applications. J. Mater. Sci. Mater. Electron. 31, 18278–18288 (2020)
Yetim, N.K., Aslan, N., Koç, M.M.: Structural and catalytic properties of Fe3O4 doped Bi2S3 novel magnetic nanocomposites: p-nitrophenol case. J. Environ. Chem. Eng. 8(5), 104258 (2020)
Kavousi, F., Goodarzi, M., Ghanbari, D., Hedayati, K.: Synthesis and characterization of a magnetic polymer nanocomposite for the release of metoprolol and aspirin. J. Mol. Struct. 1183, 324–330 (2019)
Tumturk, H., Sahin, F., Turan, E.: Magnetic nanoparticles coated with different shells for biorecognition: high specific binding capacity. Analyst. 139(5), 1093–1100 (2014)
Hedayati, K., Goodarzi, M., Kord, M.: Green and facile synthesis of Fe3O4-PbS magnetic nanocomposites applicable for the degradation of toxic organic dyes. Main Group Metal Chem. 39(5-6), 183–194 (2016)
Lwin, N., Fauzi, M.A., Sreekantan, S., Othman, R.: Physical and electromagnetic properties of nanosized Gd substituted Mg–Mn ferrites by solution combustion method. Phys. B Condens. Matter. 461, 134–139 (2015)
Ceylan, A., Ozcan, S., Ni, C., Shah, S.I.: Solid state reaction synthesis of NiFe2O4 nanoparticles. J. Magn. Magn. Mater. 320(6), 857–863 (2008)
Nawale, A.B., Kanhe, N.S., Patil, K., Bhoraskar, S., Mathe, V., Das, A.: Magnetic properties of thermal plasma synthesized nanocrystalline nickel ferrite (NiFe2O4). J. Alloys Compd. 509(12), 4404–4413 (2011)
Jacintha, A.M., Umapathy, V., Neeraja, P., Rajkumar, S.R.J.: Synthesis and comparative studies of MnFe 2 O 4 nanoparticles with different natural polymers by sol–gel method: structural, morphological, optical, magnetic, catalytic and biological activities. J. Nanostruct. Chem. 7(4), 375–387 (2017)
Amighian, J., Mozaffari, M., Nasr, B.: Preparation of nano-sized manganese ferrite (MnFe2O4) via coprecipitation method. Phys. Status Solidi C. 3(9), 3188–3192 (2006)
Aslibeiki, B., Kameli, P., Ehsani, M.: MnFe2O4 bulk, nanoparticles and film: a comparative study of structural and magnetic properties. Ceram. Int. 42(11), 12789–12795 (2016)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rezaei, M., Mirkazemi, S.M. & Alamolhoda, S. The Role of PVA Surfactant on Magnetic Properties of MnFe2O4 Nanoparticles Synthesized by Sol-Gel Hydrothermal Method. J Supercond Nov Magn 34, 1397–1408 (2021). https://doi.org/10.1007/s10948-021-05830-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-021-05830-0