
Abstract
Nano-crystalline ferrites with the chemical formula MFe2O4 (M = Zn, Ni, Cu, and Co) were synthesized via autocombustion route with citric acid as fuel. The ratio of citric acid to metal nitrate was taken as 1:1. The synthesized samples were characterized by powder X-ray diffraction and far-IR spectroscopy. The measured lattice constants and observed characteristic IR absorption bands of all samples were in good agreement with the reported values and showed the formation of a cubic spinel structure. The crystallite sizes of all samples were determined using high-intensity peaks and a W-H plot. The crystallite sizes of all samples were observed to be in the range of 16–26 nm with the least crystallite size of 16.8 nm for copper ferrite. All structural parameters were calculated using an experimental lattice constant and an oxygen positional parameter and correlated with far-IR results. Zinc and copper owing to their large cation radii showed the expansion of the lattice in view of the oxygen positional parameter. Magnetic measurements were carried out using a vibrating sample magnetometer at room temperature, and parameters such as saturation magnetization, coercivity, remanence, squareness ratio, and Bohr magnetons were calculated. Among all synthesized nano-ferrites, cobalt nano-ferrite showed the highest saturation magnetization of 41 emu/g, whereas zinc ferrite displayed a low saturation magnetization value of 12.87 emu/g.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Ferrites crystallize into a spinel structure with the general chemical formula AB2O4. The spinel structure is determined by bonding and arrangement of the oxygen ions in the lattice. The elementary unit cell of a spinel lattice contains 8 molecules of AB2O4 in which 32 oxygen ions form a cubic closepacked structure thereby providing 96 available interstitial sites. Among these 96 interstitial sites, 64 are tetrahedral (A) and 32 are octahedral (B) sites; A and B sites are surrounded by 4 and 6 oxygen ions respectively. There are 32 oxygen atoms which are in direct contact forming a closepacked facecentered cubic structure and these sites are only partially occupied by 24 metal ions. Amongst these, 8 are divalent and 16 are trivalent cations. According to the distribution of cations, these are categorized into normal, inverse and mixed spinel ferrites: In the normal type of spinel ferrites the tetrahedral sites are occupied by only the divalent ions and the octahedral sites are occupied by the trivalent ions only. These ferrites show poor magnetic properties. Examples of such ferrites are zinc ferrite ZnFe2O4 and cadmium ferrite CdFe2O4. In inverse spinel ferrites 8 out of 16 trivalent ions occupy tetrahedral sites and the octahedral sites are occupied both by divalent ions and the remaining trivalent ions. Nickel ferrite NiFe2O4 fall under this category. For mixed ferrites, both A and B sites are occupied both by divalent and trivalent ions. Examples are nickel-zinc, cobalt-zinc and copper-zinc ferrites.
The remarkable and fascinating properties of ferrites made them stood in front of all materials used in electromagnetic applications. The spinel ferrites are very attractive among them. The simultaneous display of outstanding magnetic and electric properties of these materials in nano-scale made them even better than their bulk counterparts. Among commercially available ferrites, cubic spinel ferrites are one of most technologically important on account of their applicability in different areas like absorbent materials (from microwave to radio-frequencies), high-density magnetic recording media, magnetic-core materials, and even in biomedicine [1]. Storage media is one of the technically emerging, and there is continuous demand for these materials; several attempts were made on nano-ferrites to make them suitable for the said application. The ideal characteristics for data storage media are the following: a ferrite should posses magnetization as high as possible, coercivity high enough to 600 Oe, and a narrow particle distribution with single domain size (remanance ratio around 0.5) [2].
The properties of spinel ferrites strongly depend on the method of preparation because the behavior of the system can be modified by tuning the particle dimension as well the distribution. Especially in the nanoregime, surface effects play a crucial role in determining the performance of the material in electric and magnetic fields [3]. The cation distribution shows remarkable change when the system changes from bulk to nano-form. Extensive work has been reported in the field of nanoferrites synthesized in different routes and with different choices of cationic substitutions. Though some of them are successful, work can still be extended on reproducibility of the compound. The most popular methods for synthesizing these materials are highenergy ball milling, co-precipitation, sol-gel, combustion and forced hydrolysis etc. Previously, moderate research reports are available with simple ferrites (MFe2O4) synthesized in these routes [2, 4,5,6,7]. Among the available synthesis procedures, the citric acid autocombustion method showed better quality of the ferrite as it produces a singlephased material at the end of the reaction without additional heat treatment. Furthermore in this method there are many possibilities to modify the reaction and optimal conditions to attain the desired particle size and time for the completion of the reaction [1].
The aim of this present research work is to investigate the basic structural and magnetic properties of simple nanoferrites MFe2O4 with a cation (M) as diamagnetic (zinc), paramagnetic (copper and nickel) and ferromagnetic (cobalt) substitutions prepared via the autocombustion method with citric acid as fuel with less time for completion of the reaction. The investigation of these ferrites may provide a complete understanding of the behavior of these cations in the tetrahedral (A-site) and octahedral (B-site) sub-lattices of ferrite structure at full concentration of single divalent metal cation and also facilitate the use of different combinations of these cations in a single ferrite system The influence of these cations on the magnetic properties is discussed to the maximum extent possible, in view of structural modifications resulting in the strength weakening of inter and intramolecular interactions for potential data storage applications
2 Experimental Procedures
2.1 Synthesis
High-purity raw materials, zinc nitrate Zn (NO3)2 ⋅6H2O, nickel nitrate Ni (NO3)2 ⋅6H2O, copper nitrate Cu (NO3)2 ⋅3H2O, cobalt nitrate Co (NO3)2 ⋅6H2O, iron nitrate Fe (NO3)3 ⋅6H2O, and citric acid (C6 H 8O7 ⋅H 2O), were taken in stoichiometric proportions and dissolved each material separately in a minimum amount of de-ionized water. Thus formed cationic solutions are mixed intimately and stirred for an hour to improve the homogeneity. Then, an aqueous solution of citric acid in measured quantities (molar ratio of metal nitrates to citric acid as 1:1) was added slowly to the precursor. This mixture was stirred for 30 min until it turned into a clear brownish solution, and this was heated around 65 °C under continuous stirring. Slowly, the temperature is raised up to 100 °C and it is continued beyond to 135 °C and kept constant. Initially, water was slowly evaporated and as the time proceeds, viscous liquid was formed leading to the gelation. The internal temperature of the gel was slowly raised and proceeded with burning of the gel resulted in the formation of chunks of fluffy product by the releasing of brownish gases. The entire reaction was completed in about 2 h time. After the completion of the reaction, these flakes were grounded for 1 h (both wet and dry) to achieve fine powder using agate mortar and pestle.
2.2 Characterization Techniques
2.2.1 Powder X-ray Diffraction
In order to confirm that the process adopted was the correct one and prior to further analysis, the powder X-ray diffraction technique was performed to identify the structure of the material. A BRUKER D8 ADVANCE POWDER X-ray diffractometer with Cu k α (λ = 1.5406 Å) radiation was used to characterize the samples.
2.2.2 Far-IR Spectroscopy
Infrared measurements were recorded using a PERKIN ELMER–FTIR spectrometer; it has a resolution of 1 cm−1 and scan range of 700 to 50 cm−1 at room temperature using a KBr pellet.
2.2.3 Vibrating Sample Magnetometer
Magnetic measurements were performed using a LAKESHORE 7400 vibrating sample magnetometer up to 15,000 Oe at room temperature. Saturation magnetization, coercivity, remnant magnetization and other related parameters are obtained from the recorded MH curves data.
3 Results and Discussion
3.1 Structural Studies
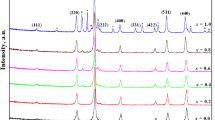
For simplicity, the present series of samples are named as ZnF (zinc ferrite), NiF (nickel ferrite), CuF (copper ferrite), and CoF (cobalt ferrite). The X-ray diffraction patterns of all ferrite samples confirmed the single-phase spinel structure (Fig. 1). All peaks were assigned to known planes of cubic spinel, according the standard JCPDS cards [22-1086, 34-0425]. In XRD, the pattern of copper ferrite showed some addition peaks, which were represented with a dot over the peaks. They are unidentified and may be due to few amounts of copper oxide not entering the lattice.

Powder X-ray diffraction pattern of simple ferrites MFe2O4 (M = Zn, Ni, Cu, and Co)
The inter-planar spacing d of different crystal planes has been obtained from the intensity versus 2 𝜃 graph of the X-ray diffraction pattern using Bragg’s law [8],
Inter-planar distances calculated from the glancing angle for all samples are presented in Table 1, and they are slightly smaller than the reported values which accounted for less crystallite size [1]. As per d-spacing of the CuF, it is very much clear that it belongs to cubic spinel and there is no deviation from ideal d-spacing values, which demonstrates that the tetragonal phase was not present. The plane indices (h k l) have been assigned to each peak using the X-ray data of the standard spinel structure. The lattice parameter a has been calculated from each peak using equation \(a=d \sqrt {h^{2}+k^{2}+ l^{2}} \) [7]. The so-obtained lattice constant values were extrapolated with Nelson-Riley function F(\(\theta )=\frac {1}{2}\left (\frac {{\cos }^{2}\theta } {\sin \theta } +\frac {{\cos }^{2}\theta } {\theta } \right )\) in a graph [8] for accurate determination of lattice parameters. Lattice constants corresponding to high-angle planes were considered for the Nelson-Riley plot. The lattice constant and inter-planar spacings of different crystal planes for the four samples are shown in Table 1.
The lattice constant values were in accordance with cationic radii and obeyed Vegard’s law. Copper being larger in size, still CuF showed less value, which is related to the low crystallite size of the system. The measured values are in good agreement with the reported values [1, 9], indicating the entering of all cations into the respective spinel lattices.
3.2 Determination of Average Crystallite Size
The crystallite size D for as-prepared samples has been calculated from the experimentally observed broadening (full width at half maximum (FWHM)) of the most intense (311) peak of the X-ray diffraction pattern using the Scherrer equation [10],
where k is the Scherrer or shape factor (The Lorentzian and Gaussian limits for the shape factors are 0.6366 and 0.9394 respectively [10]), λ is the wavelength of X-rays (Cu-K α radiation = 1.5406 Å), 𝜃 is the Bragg angle, and from Warren’s formula β = β e − β o, β e is the FWHM of the diffraction peak in radians and β o instrumental broadening.
The accurate average crystallite size for as-prepared samples was determined using the Williamson-Hall method. Accordingly, the broadening of the X-ray diffraction peak is believed to be due to the crystallite size of the samples and their strain or lattice distortion; mathematically, this can be written as [10]
In practice, this equation can be expressed as \(\frac {\beta \cos \theta } {\lambda }{=}\frac {k}{D} + \frac {4\varepsilon \sin \theta } {\lambda }\)
-
β = experimentally observed FWHM
-
β size = FWHM due to the effect of crystallite size only
-
β strain = FWHM due to the effect of micro-strain only
-
ε = lattice strain
-
𝜃 = incident angle of the X-ray
-
λ = wavelength of the X-ray beam
By using this relation, contributions from crystallite size and micro-strain can be separated in a straightforward approach by fitting the experimentally observed diffraction peak data. In general, the peaks are either Lorentzian or Gaussian or may be voigt shaped. In the present series of simple ferrites, the shape of the curve fitted for the most intense diffraction peak (311) was found to be either Gaussian or Lorentzian. Separation of size and strain components can be done by plotting β cos 𝜃/ λ versus sin 𝜃/ λ (Williamson-Hall plots). If size broadening is the only significant contribution to peak width, the plot gives a horizontal line. If strain broadening is also an important contribution, the plot is a linear function of sin 𝜃. Williamson-Hall plots for all as-prepared ferrite compositions were drawn, and the average crystallite size was calculated from the y-intercept of the plots by making strain component zero. The profiles of (311) peak after curve fitting along with W-H plots of all samples are shown in Fig. 2. The crystallite size and the strain values are presented in Table 2.

W-H plots for all samples (profile fits of (311) are shown as insets)
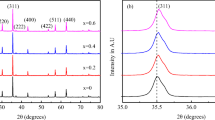
Prior to the W-H plot, FWHM values of the most prominent peaks were calculated for all samples. Using these values, crystallite sizes were obtained and they were in the range of well below 28 nm and confirming the formation of nanocrystalline ferrites. These crystallite sizes of different peaks for each ferrite showed a little variation which would be a sign of narrow distribution of particles. The crystallite sizes from W-H plots were slightly smaller than those obtained from the (311) peak alone. This is a feature of the W-H plot, in which size broadening of the peak only considered for crystallite size determination. Out of all samples, CuF showed the least crystallite size of 16.8 nm and CoF the highest value of 25.5 nm. All samples showed positive lattice strain indicating a small elastic expansion of the structure. The positive lattice strain indicates that it was a tensile strain. ZnF and CoF showed high value of strain attributed to large sizes of zinc and cobalt ions, which is displayed in terms of the shifting of diffraction peaks towards lower 2 𝜃 values. Nickel and copper showed less strain due to smaller radii and crystallite size. These changes were reflected in the displacement of the (311) peak. In the present series of samples, all cations “M” are divalent, but they have different masses, radii, site preference energies, and number of free electrons. This can be clearly understood by observing the X-ray diffraction pattern, and for clear illustration, (311) peak of X-ray diffraction peaks of all samples is shown in Fig. 3.

(311) peak of the XRD pattern of the present series of nanoferrites
The properties of a nanoferrite vary noticeably depending upon the distribution of the cations among tetrahedral and octahedral sites of spinel lattice. The factors which can influence the distribution of the metal ions over the A and B sites are (1) their ionic radii and electronic configuration of the metal cation, (2) the electrostatic energy of the spinel lattice, and (3) particle size [8, 11].
One of the various methods that has been employed to study the cation distribution is the calculation of intensities of X-ray diffraction peaks. Cation distribution is proposed on the basis of intensity calculations and it was verified using the theoretical lattice constant.
In order to evaluate theoretical intensities of the diffraction peaks, the following formula was used as suggested by Buerger [12]:
where I h k l is the relative integrated intensity, F h k l is the structure factor, P is the multiplicity factor, and L P is the Lorentz polarization factor, which is expressed as L P = [(1 + cos2 𝜃)/sin2 𝜃 cos 𝜃].
Among all observed reflections in X-ray diffraction patterns, the planes with the Miller indices (220), (400), (422), and (440) were considered for intensity calculations, since these are considered to be structure-sensitive planes. Out of these four planes, (220) and (422) are sensitive to any change of cations on the A-site while (400) and (440) are sensitive to cations on both sites [13]. Amongst the available diffraction intensities of different planes, (311) plane is nearly independent of the oxygen parameter and the cation distribution will not change significantly with such changes. Therefore, the intensity of (311) peak was taken as 100% [13]. All integrated intensities of the four planes under consideration were estimated with reference to (311) plane.
The structure factors of different crystal planes were calculated by using the following equations mentioned below [13].
where F A, F B, and F o are structure factors of A-site and B-site cations and oxygen atoms. The structure factor F is the sum of scattering factors of individual ions present in that site. The atomic scattering factor, defined as the ratio of amplitude of the wave scattered by all atoms to amplitude scattered by free electrons for the same incident beam. The intensities of the diffracted beams from the crystal planes depend on the group of atoms in the unit cell and scattering power of these atoms [13]. In these set of theoretical X-ray diffraction intensities, the ratios of integrated intensities were estimated by neglecting the Debye factor and the temperature factors [14]. Furthermore, the orientations of crystal planes of the sample will not affect the ratios of intensities, which is allowed to consider intensity ratios rather than individual intensities.
All theoretical intensity ratios were calculated by changing cations on either site. From these intensity ratios, the cation distribution which was the closest match with experimental values is presented in Table 2. Slight difference was noticed in calculated and observed intensities, and it is owing to the atomic scattering factors, which are derived from the electron contributions of atoms at rest, i.e., at absolute zero. Whereas, the X-ray diffraction patterns of all samples are recorded at room temperature; at ordinary temperatures, thermal vibrations of atoms will cause them to occupy a large volume than they would at rest, making the scattering still smaller. Although for all intensity ratios a minimum difference in theoretical and experimental ones is achieved, the intensity ratio I(400)/I(220) showed a larger difference. The reasons for this observation are answered with the following considerations. The presence of cations in different oxidation states (for example, formation of Fe2+ ions as well as Cu1+ ions in copper ferrite due to oxygen ion vacancies) has different scattering factors which are not taken into consideration during calculations. This is an unlikely reason in nanoferrites. Moreover, the Buerger equation is based on the assumption that it was applicable for the specimens having random orientations in space. However, all polycrystalline specimens will show preferential orientation of the grains in more or less manner. The method executed here was based on the assumption that the crystal is an ideally imperfect crystal; however, in practice, during the crystal growth and especially in nanoregime, some vacancies will be created. Therefore, crystal may consist of some or all accurate parallel planes which may be nearly perfect and have low refracting power. This would be the cause for observed intensity differences for the specific planes. These can be verified by vacancy model in spinel ferrite, which is discussed in the next section. The proposed cation distribution is presented as shown in Table 3.
Structural aspects of spinel ferrites play a crucial role in determining the magnetic properties of ferrites such as magnetization, remanence, etc. After arriving at a reliable cation distribution, a detailed study was performed on structural parameters and those were correlated with the data obtained from the far-infrared spectrum.
The mean radius of the tetrahedral site is given by
The mean radius of the octahedral site is given by
where r M(A), r M(B),r Fe(A), and rFe(B) are the radii of metal cations (Zn, Ni, Cu, and Co) and iron ions at A and B sites respectively (radii of cations at tetrahedral site—zinc (0.6 Å), nickel (0.55 Å), copper (0.57 Å), cobalt (0.58 Å), iron (0.49 Å) and for octahedral sites—zinc (0.74 Å), nickel (0.69 Å), copper (0.73 Å), cobalt (0.745 Å), and iron (0.645 Å) ions in the octahedral site respectively [15]).
From the cation distribution, it was clearly shown that all cations have octahedral occupancy, especially nickel, copper, and cobalt. Thus, more iron ions were migrated to tetrahedral sites; as such, the effective ionic radii of iron reduced from 0.645 to 0.49 Å. In view of this, r A values are much lower than r B. Expansion/contraction of lattice depends on the movement of cations between the available sub-lattices. Since all samples showed positive lattice strain, the presence of tensile strain is obvious. This indicates that there was a slight movement of anions surrounding the cations. The movement of oxygen ions can be explained by means of oxygen positional parameter u. The oxygen positional parameter can be calculated by assuming the origin at A-site or at B-site. Since A-site cation belongs to \(\overline 4 \mathrm {3m}\) symmetry group, the u parameter measured by considering origin at A-site, is denoted by \({ u^{\mathrm {43m}}}_{}\). Since, the B-site cation belongs to the \(\overline 3 \mathrm {m}\) symmetry group, the u parameter measured by considering origin at B-site, is denoted by u 3m [14]. In the present series of samples, the oxygen positional parameter u was measured by assuming the center of symmetry at (3/8, 3/8, 3/8) or origin at A-site (\(\overline 4 \mathrm {3m}\) symmetry group), also represented as u 43m. The oxygen parameter can be calculated from known r A and r B values. The following formula is used to convert the center of symmetry from (1/4, 1/4, 1/4) to (3/8, 3/8, 3/8) or vice versa, u 43m = u 3m + (1/8). The ideal values of u 43m = 0.375 and u 3m = 0.25. The values of u 43m and u 3m and calculated lattice constants are shown in Table 4. It is obvious from Table 4 that both u values were slightly larger than ideal values, which would be the evidence that the structure is slowly coming to the ideal case; oxygen ions were moving away from tetrahedral coordinated cations along < 111 > direction. This resulted in the expansion of the tetrahedral site while the octahedral site contracts. This anion dilation happens with the variation in u, and this has no effect on the symmetry of the octahedra, while the symmetry of the tetrahedra will be affected. This expansion of sub-lattice(s) was attributed to the movement of oxygen ions without changing the structure symmetry of the unit cell as a whole. Based on the fact that cations in each sample were entering both sites at different concentrations along with the movement of Fe3+ ions, it is thus quite possible to observe changes in structural parameters like bond lengths; shared and unshared edges, etc. were viewed on the basis of the oxygen positional parameter (u).
The mean inter-ionic distances (bond lengths) at tetrahedral (r t) and octahedral sites (r o) were estimated using the following relations [14]:
The tetrahedral edge length d AE, the shared octahedral length d BE, the unshared octahedral edge d BEu, and the inter-ionic distances (bond lengths) at tetrahedral (d AL) and octahedral sites (d BL) of the present nanoferrites have been calculated using the following equations, and they are represented in Table 4.
where δ represents the deviation from the oxygen parameter, δ = u 3m − 0.375.
Based on the proposed cation distribution, the theoretical lattice constant [13] is calculated by using the relation
Here, R o is the radius of the oxygen ion (1.38 Å).
The theoretical and experimental lattice constants are in agreement except copper showing slight variation. The low values of a th are due to few concentrations of CuO not entering the lattice or the possible formation of Fe2+ ions in the samples, and the estimation is based on Fe3+ ions. The change of sub-lattice symmetries is reflected in bond angles. Since the strength/magnitude of super-exchange interactions are directly proportional to bond angles and inversely proportional to bond lengths, it is considered necessary to mention the variations in these parameters with composition. The changes in lattice constant and oxygen parameter can be expressed in terms of cation-cation and cation-anion bond lengths and bond angles. The cation-cation (M-M) and cation-anion (M-O) bond distances and the angles between these inter-ionic distances (bond angles) were estimated using the simplest trigonometric formulae [14].
From Table 4, it is clear that all samples displayed the standard bond angles [1, 14] observed in ferrites. ZnF showed slightly larger values compared to remaining systems, showing the weakening of exchange interactions. The CuF system showed similar angles with the rest of the samples, and no major changes were observed in all structural characteristics indicating the cubic spinel nature and tetragonal nature at higher concentrations of copper were not observed in this case. Among all cation-cation bond distances, b values are comparatively small, because there are no intervening anions to obstruct neighboring cations. From Table 4, it is clear that there are commonly occurring slight deviations observed in all angles from the ideal bond angles. But the zinc substitution produced more pronounced variations in the bond angles, showing that there was a possibility of slight alteration in the alignment of the cations present in the lattice sites (spin canting). In order to investigate the structural deviations in detail, vacancy model calculations [19] were carried out to know the occupancy of ions in the lattice.
Ionic packing coefficients at tetrahedral and octahedral sites are \(P_{\mathrm {A}}=\frac {r_{\mathrm {t}}}{r_{\mathrm {A}}}\) and \(P_{\mathrm {B}}=\frac {r_{\mathrm {o}}}{r_{\mathrm {B}}}\)
where r t and r o are the interstitial radii and r A and r B are the average values of the ionic radii at the tetrahedral and octahedral sites, respectively.
Vacancy parameter \(\beta =\frac {\mathrm {(}a_{\text {th}}^{3}\mathrm {-}a_{\exp }^{3}\mathrm {)}}{a_{\text {th}}^{3}}\mathrm {\ast 100}\)%
The values of P A and P B were nearly equal to 1, demonstrating that there were very few possible cation/anion vacancies present in the samples. The same was displayed in the vacancy parameter also. Vacancy model calculations indicate that all samples displayed characteristic values of nanoferrites, except CuF had moderate vacancies of ions may be attributed to the few amounts of CuO not entered the lattice interstitial sites leading to some vacancies, the same is demonstrated in theoretical lattice constant which is larger than experimental value as well as displayed additional peak in X-ray diffraction pattern.
3.3 Infrared Studies
The infrared vibrational spectrum of a molecule is considered to be a fingerprint for a molecule, and it is a unique physical characteristic of the molecule. As such, using this characterization tool for identification of the structure by comparing the spectrum of “unknown” molecule with previously recorded reference spectra. Spinel ferrites crystallize in cubic form with a space group Fd3m-\(\mathrm {O}_{\mathrm {h}}^{7}\). According to Waldron [20], ferrites are considered to be continuously bonded crystals. As far as spinel ferrites are concerned, the active absorption region is observed to be 100–800 cm−1. The position of absorption bands in this region changes with the composition of the ferrite. The infrared spectrum of spinel ferrites in the nano-regime depends on the method of preparation, size of the particles/grains, cation radii, and atomic mass. Group theory based calculations along with space group and point symmetry considerations suggest that any (normal and inverse) cubic spinel ferrite should exhibit four infrared active absorption bands designated as υ 1, υ 2, υ 3, and υ 4. The first three bands are observed due to tetrahedral and octahedral metal complexes while the fourth one is due to some type of lattice vibrations [21]. These characteristic bands are lattice vibrations which strongly depend on any structural changes brought about by a substituent or incorporated metal cation. Based on symmetry considerations, these four fundamental absorption bands are due to T2 and E vibrations of symmetry [21]. Absorption bands υ 1 and υ 2 are assigned to the intrinsic lattice vibrations of E-type symmetry, while the rest of the bands (υ 3 and υ 4) also are assigned to lattice vibrations having T-type symmetry. These types of atomic displacements T and E are due to bond bending and bond stretching so that in general υ(T2) < υ(E). Moreover, most of the ferrites show a mixed nature, so there are two types of metals in majority of the tetrahedral and octahedral sites (M2+ and Fe3+) in each sample. Therefore, four types of phonon modes Eo, Et, To, and Tt can be observed, where the superscripts o and t indicate the atomic displacements around octahedral and tetrahedral sites involving metal ions and oxygen ions in a normal ferrite corresponding to the E and T symmetry. The order of the absorption bands is given by the following expression for any ferrite system whether it is normal, mixed, or inverse is υ 4 (Tt) < υ 3 (Tt/o) < υ 2 (Eo) < υ 1 (Et). Out of these four, the υ 4 band frequency depends on the mass of the tetrahedral cation and is assigned to some type of vibrations involving tetrahedral metal ions. However, the site symmetry of the metal complex and site preference of the divalent metal cation influences the band υ 3 in a mixed or inverse ferrite; as such, υ 3 is considered to be from the octahedral metal complex [22]. The far-IR spectrum of all samples shown in Fig. 4.

Far IR spectrum of simple ferrites MFe2O4 (M = Zn, Ni, Cu, and Co)
In the present, infrared spectra of all four ferrites showed the characteristic ferrite absorption bands, confirming the formation of the cubic spinel structure. Among the four ferrite systems, nickel and cobalt ferrites displayed similar absorption bands, which are correlated to their common preference towards octahedral sites and very close atomic masses (Ni 58.693, Co 58.933). In these two systems, out of four characteristic absorption bands, υ 1 and υ 2 are having high intensity since the motions involved are predominantly those of the oxygen ion, indicating the strength of Fe3+–O2− bonds at tetrahedral and octahedral sites (evident from force constants) as well as the concentrations of iron ions in the system. The positions of the absorption bands are well in agreement with the reported values [23, 24]. Copper ferrite and zinc ferrite samples showed four characteristic absorption bands. Amongst υ 1 is not very sharp; instead, it is very broad indicating a reasonable amount of tetrahedral occupancy of divalent metal cations. Both these ferrites showed mixed ferrite nature. Especially, in ZnF, zinc ions do not have free electrons and these are equally distributed between the two sites resulted in the lowering of the mean ionic charge thereby decreasing force constants. Due to this fact, the mean tetrahedral bond length would increase [25]. The same is observed in structural characteristics discussed in the previous section. ZnF showed three characteristic bands, which are in very close agreement with the reported values [26]. The force constants of Me-O bonds at octahedral and tetrahedral sites of all samples were calculated using relations [19] and they displayed the same order, but the zinc ferrite sample showed slightly lower values (Table 5). This was reflected in the bond distances, and the effects are also shown in saturation magnetization, which is discussed in the next section.
3.4 Magnetic Studies
The magnetization response of all ferrites at 15,000 Oe and at room temperature is shown in Fig. 5. Since at a maximum field of 15,000 Oe the magnetization has not reached a saturated value, therefore, the law of approach to saturation [27] is applied to obtain saturated values of all samples and a graph showing M versus 1/H for CoF is shown in Fig. 5.

Variation of magnetization of all samples MFe2O4 (M = Zn, Ni, Cu, and Co) with applied field and law of approach to saturation graph for cobalt ferrite
In general, the magnetization of bulk ferrites is due to cationic arrangement on both sites (sub-lattices) of the spinel lattice and super exchange interactions and it can be explained by Neel’s two-sub-lattice model [JMMM], according to which a magnetic ferrite crystal has a domain structure similar to that of ferromagnetic metals. Accordingly, the magnetization of each domain is associated with the magnetic moments of the constituent ions. In view of the contribution to the magnetic moment from the individual ions present in the ferrite, the oxygen ions have no magnetic moment, since the free oxygen atom has a partially unfilled 2p-sub-shell which gets filled on acquisition of two electrons and becomes O2− negative ion. When the free oxygen converts to O2− negative ion, this makes no direct contribution to the magnetic moment of the domain. The magnetic moments of the remaining ions arise from the parallel uncompensated electron spins in the unfilled outer sub-shells of the cations only. As in ferromagnetic metals and in paramagnetic solids, internal fields quench the moments due to the orbital motion of the electrons. Among the available cations, iron ions have a major role in the magnetization of ferrites since they are the largest in number of the ferrite system. In ferrites, it is possible for the iron ions to present in both + 2 and + 3 states. In case of divalent iron ion, there are four uncompensated spins in the 3d sub-shell, so that the iron atom, assuming quenched orbits, has a magnetic moment of 4 μ B (μ B being the Bohr magneton) due to spin. The divalent iron ion having it by losing two electrons from the 4s shell has a moment of 4 μ B. On becoming the trivalent ion, it loses an additional electron from the 3d sub-shell increasing the uncompensated electron spins to 5 and the resultant moment becomes 5 μ B. Between the spinning electrons in the neighboring metal ions, strong quantum mechanical forces of interaction occur. Based on this assumption, Neel developed a theory, according to which there exist three kinds of exchange interactions in ferrites: the interaction between the various magnetic ions located at A-site (AA interaction), the interactions between the various magnetic ions located at B-site (BB interaction), and the interaction of magnetic ions at A-site with those at B-site (AB interaction); out of these, AB interactions are the strongest and in which the alignment of all the magnetic spins at A-site in one direction and those at B-site in the opposite direction. The net magnetic moment of the lattice is therefore the difference between the magnetic moments of B and A sub-lattices, i.e., M = MB − MA.
However, in the case of nanoferrites, the saturation magnetization of the system can be explained not only by Neel’s theory but also on the (1) method of preparation, (2) crystallite size or particle size, (3) coercivity, (4) anisotropy, (5) alignment of sub-lattices (spin canting effects), and (6) thermal treatment [8, 28].
M-H curves of all samples were recorded at room temperature and at 1.5 kOe are presented in Fig. 6. In ZnF, the presence of a more diamagnetic ion concentration on either side of the sub-lattices decreases the strength of interaction between A and B, thereby decreasing the magnetization. As evident from the structural properties like bong lengths, angles, etc., the equal occupancy of larger zinc ions on either site leads to magnetic dilution and structural deformation, thus producing a strong effect on the magnetic properties. The poor magnetic properties of zinc ferrite are also attributed to the spin canting effect introduced by the zinc ions present at the octahedral site. In this sample, half of the zinc ions occupied the octahedral site which changes with heat treatment and particle size. It is very much familiar in nanozinc ferrites [1]. Though copper is having only 1 μ B of magnetic moment, the CuF sample showed nearly the same magnetic characteristics as NiF except in magnetization, which is due to the ability of copper to diffuse through the spinel lattice. On the other hand, NiF has a slightly high M s value, since nickel has a magnetic moment of 2 μ B. As it is evident from the literature, CuF also has a slight structural deformation due to its large radius and at higher concentrations leading to spin canting effects. The low values of magnetization of NiF and CuF compared to CoF were attributed to less number of free electrons as well as reduction in particle size. Among the four simple nanoferrites of the series, CoF showed the highest magnetization, coercivity, and remanence. The strong ferromagnetic nature of the cobalt ion with 3 μ B is responsible for this high value. The coercivity is very high compared to the same system synthesized in different routes, which is attributed to the anisotropic nature of cobalt at the octahedral site which is not able to quench the orbital magnetic moment leading to strong L-S coupling [1]. Moreover, the crystallite size of the as-synthesized CoF was observed to be 25.5 nm and it is very close to the critical size of 25 nm, at which cobalt ferrite displayed a high coercivity value reported in the literature [29]. All of the simple nanoferrites presented here showed slightly higher saturation magnetization compared to the reported values [1, 9, 23, 29]. All divalent cations except zinc showed octahedral site preference, which comes out to be the reason for higher magnetization values.

M-H curves of all samples MFe2O4 (M = Zn, Ni, Cu, and Co) recorded at room temperature
Experimental Bohr magnetons per one formula unit are calculated from the following relation [8]:
where M wt is the molecular weight and M s is the saturation magnetization, respectively.
The experimental Bohr magneton values of CoF and NiF are half of the theoretical values, since theoretical Bohr magneton values were calculated at 0 K. Whereas, in CuF and ZnF samples, the experimental Bohr magneton is less than half of the theoretical ones, which is attributed to weak exchange interactions and spin canting effects. The low values of experimental Bohr magnetons observed for the present as-prepared samples were also attributed to low density of the samples because of the absence of thermal treatment. The values of squareness of the loop or the remanence ratio are observed to be around 0.26–0.38 showing that these particles are approaching a limiting value 0.5 which corresponds to noninteracting single domain particles [1, 29]. The values of saturation magnetization, coercivity, remanence, squareness ratio, and experimental and theoretical Bohr magnetons are shown in Table 6.
4 Conclusions
Simple nanoferrites are synthesized by the citric acid autocombustion method. This method produced single-phased nanocrystalline material confirmed by powder X-ray diffraction. The lattice constants of present nanoferrites were in good agreement with the reported values. Cation distribution was proposed from XRD intensity rations, and in all ferrites, divalent cations were found to be occupied in both tetrahedral and octahedral sites. Out of these, nickel, copper, and cobalt ions showed strong octahedral site preference. A far-IR study showed the characteristic absorption bands, and the band positions showed that divalent cations are distributed on both sites. The force constants of ZnF were low in value, showing the change of bond strengths with a nonmagnetic ion in both sites. The octahedral occupancy of zinc ions showed its strong influence on structural properties of ZnF. The M-H loops of all ferrites showed that the samples were not saturated even at a high field of 15,000 Oe. Among the four systems, NiF and CuF showed similar structural and magnetic properties. CoF displayed its strong ferromagnetic nature with a high coercivity and remanence. Among the four nanoferrites, NiF and CuF displayed ideal characteristics for data storage media, as they displayed moderate magnetization, coercivity high enough to 600 Oe (which is around 500 Oe), and squareness ratio near 0.5.
References
Raut, A.V., Barkule, R.S., Shengule, D.R., Jadhav, K.M.: Synthesis, structural investigation and magnetic properties of Zn2 + substituted cobalt ferrite nano particles prepared by the sol–gel auto-combustion technique. J. Magn. Magn. Mater. 358–359, 87–92 (2014)
Ashiq, M.N., Naz, F., et al.: Role of Co-Cr substitution on the structural, electrical and magnetic properties of nickel nano-ferrites synthesized by the chemical co-precipitation method. Mater. Res. Bull. 47, 683–686 (2012)
Kumar, V., Rana, A., Yadav, M.S., Pant, R.P.: Size-induced effect on nano-crystalline CoFe2O4. J. Magn. Magn. Mater. 320(11), 1729–1734 (2008)
Atif, M., Nadeem, M., et al.: J. Alloy. Compd 509, 5720–5724 (2011)
Chaoquan, H., Gao, Z., Yang, X.: One-pot low temperature synthesis ofMFe2O4 (M = Co, Ni, Zn) superparamagnetic nanocrystals. J. Magn. Magn. Mater. 320(8), L70–L73 (2008)
Huo, J., Wei, M.: Characterization and magnetic properties of nanocrystalline nickel ferrite synthesized by hydrothermal method. Mater. Lett. 63(13–14), 1183–1184 (2009)
Yanez-Vilar, S., Sánchez-Andújar, M., Gómez-Aguirre, C., Mira, J., Senaris-Rodríguez, M.A., Castro-García, S.: A simple solvo-thermal synthesis of MFe2O4 (M = Mn, Co and Ni) nano particles. J. Solid State Chem. 182(10), 2685–2690 (2009)
Sivaram prasad, M., Prasad, B.B.V.S.V., Rajesh, B.B., Rao, K.H., Ramesh, K.V.: Magnetic properties and DC electrical resistivity studies on cadmium substituted nickel–zinc ferrite system. J. Magn. Magn. Mater. 323(16), 2115–2121 (2011)
Shahbaz Tehrania, F., Daadmehr, V., et al.: Structural, magnetic, and optical properties of zinc- and copper-substituted nickel ferrite nano crystals. J. Supercond. Nov. Magn. 25(7), 2443–2455 (2012)
Maniammal, K., Madhu, G., Bijua, V.: X-ray diffraction line profile analysis of nano structured nickel oxide: Shape factor and convolution of crystallite size and micro-strain contributions. Phys. E. 85, 214–222 (2017)
Vara Prasad, B.B.V.S.: Cation distribution, structural and electric studies on cadmium substituted nickel–zinc ferrite. Mod. Phys. Lett. B 28(19), 1450155 (2014). (13 pages)
Buerger, M.J.: Crystal Structure Analysis. Wiley, New York (1960)
Vara Prasad, B.B.V.S., et al/: Structural and dielectric studies of mg2+ substituted Ni–Zn ferrite. Mater. Sci.-Poland 33(4), 806–815 (2015)
Lakhani, V., Pathak, T.K., Vasoya, N.H., Modi, K.B.: Structural parameters and X-ray Debye temperature determination study on copper-ferrite-aluminates. Solid State Sci. 13(3), 539–547 (2011)
Shannon, R.D.: Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 32, 751–767 (1976)
Parashar, J., Saxena, V.K., Jyoti, Bhatnagar, D., Sharma, K.B.: Dielectric behaviour of Zn substituted Cu nano-ferrites. J. Magn. Magn. Mater. 394, 105–110 (2015)
Maqsood, A., Khan, K., et al.: Spectroscopic and magnetic investigation of Ni-Co nanoferrites. J. Alloy. Compd. 509, 7493–7497 (2011)
Salunkhe, A.B., Khot, V.M., et al.: Low temperature combustion synthesis and magneto structural properties of Co-Mn nanoferrites. J. Magn. Magn. Mater. 352, 91–98 (2014)
Mohammed, K.A., Al-Rawas, A.D., et al.: Infrared and structural studies ofMg1−xZnxFe2O4 ferrites. Physica B 407, 795–804 (2012)
Waldron, R.D.: Infrared spectra of ferrites. Phys. Rev. 99(6), 1727–1735 (1955)
Reddy, P.V., Salagram, M.: The far-infrared spectra of mixed Mn-Mg ferrites. Phys. stat. sol. (a) 100, 639 (1987)
Josyulu, O.S., Sobhanadri, J.: The far infrared spectra of zinc ferrites. Phys. Stat. Sol., (a) 65, 479 (1981)
Lazarevic, Z.Š, Jovalekic, F., et al.: Preparation and characterization of nano ferrites. Acta Phys. Pol. A 121, 682–686 (2012)
Mund, H.S., Ahuja, B.L.: Structural and magnetic properties of Mg doped cobalt ferrite nano particles prepared by sol-gel method. Mater. Res. Bull. 85, 228–233 (2017)
Singh, R.K., Narayan, A., et al.: Thermal, structural, magnetic and photoluminescence studies on cobalt ferrite nano particles obtained by citrate precursor method. J. Therm. Anal. Calorim. 110(2), 573–580 (2012)
Pradeep, A., Priyadharsini, P., Chandrasekaran, G.: Structural, magnetic and electrical of nano crystalline zinc ferrite. J. Alloy Compd. 509, 3917–3923 (2011)
Chikazumi, S.: Physics of Ferromagnetism. Oxford University Press, Oxford (1997)
Shobana, M.K., Sankar, S.: Characterization of sol–gel-prepared nano ferrites. J. Magn. Magn. Mater. 321, 599–601 (2009)
Toksha, B.G., Shirsath, S.E., Patange, S.M., Jadhav, K.M.: Structural investigations and magnetic properties of cobalt ferrite nano particles prepared by sol-gel auto combustion method. Solid State Sci. 147, 479–483 (2008)
Acknowledgments
Authors are thankful to DST-FIST, India, for providing infrastructural facilities-equipment through No. SR/FST/PSI-194/2014 Dated: 21st July 2015.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vara Prasad, B.B.V.S., Ramesh, K.V. & Srinivas, A. Structural and Magnetic Studies of Nano-crystalline Ferrites MFe2O4 (M = Zn, Ni, Cu, and Co) Synthesized Via Citrate Gel Autocombustion Method. J Supercond Nov Magn 30, 3523–3535 (2017). https://doi.org/10.1007/s10948-017-4153-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-017-4153-y