
Abstract
The polycrystalline sample of the Ruddlesden–Popper La1.4(Sr0.95Ca0.05)1.6Mn2O7 phase was synthesized using a sol–gel method. To determine the structural and optical properties as a function of photon energy for this sample, we can use the various techniques of characterization such as X-ray diffraction, SEM, and ellipsometric spectroscopy. Based on structural analysis by Rietveld profile fitting, a single-phase sample with tetragonal I4/mmm space group was obtained. The optical properties were investigated by spectroscopic ellipsometry at room temperature in the spectral range of 300–800 nm. The optical constants: refractive index n and extinction coefficient k were also studied. These measurements revealed that the optical absorption is obtained around 590 nm and the sample has a direct transition corresponding to the energy gap which was equal to 2.25 eV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Perovskite manganites have been nowadays the subject of intense interest, both of their potential applications and a wealth of novel physical phenomena.
The lanthanum-doped perovskite manganite of the Ruddlesden–Popper series with a general compositional formula (La, A) n+1Mn n O3n+1 (A: Ca, Sr, Ba, Pb; n is the number of MnO2 layers per unit cell) is well known to have the layered-type structures [1–4]. The n = ∞ compounds of La1−x A x MnO3, having a three-dimensional (3D) crystal structure, have attracted much attention because of a variety of interesting physical properties [5]. These compounds have been investigated widely and systematically for a long time [5, 6]. The n = 2 manganite series La2−2x A 1+2x Mn2O7 crystallize with tetragonal or orthorhombic unit cell in the space group I4/mmm or Fmmm [7–10]. The homologous series contains bilayer slices of MnO6 octahedra separated by insulating rock-salt-type (La, A) O layers. These phases display two-dimensional magnetic propagation generally. It has been reported to show an anisotropic reduction in the one-electron (eg) bandwidth and exhibit some interesting electronic, magnetic, and structural properties different from those of cubic perovskites [11].
In comparison with the preparation process of La-doped single perovskites, La2−2x A 1+2x Mn2O7 materials require more complex synthesis conditions. Among the material preparation methods, a solid-phase reaction and a sol–gel method are often used for making these materials. Many works have reported the synthesis of Ln2−2x A 1+2x Mn2O7 materials using the sol–gel method because in this method, the precursors can be well mixed and the product is fine [12, 13]. Other materials employed the solid-phase reaction method, which requires high sintering temperature, long sintering time, and repeated sintering processes [14, 15].
Recently, the effects of structural, electrical, and magnetic properties with the isovalent chemical substitution of Ca2+ into the Sr2+ sites in the layered Ruddlesden–Popper perovskite have been investigated [16]. The replacement of Sr2+ ions with smaller Ca2+ ions causes an anisotropic internal chemical pressure within the compound and leads to a rich variety of magnetically ordered and electrically conductive phases [16]. However, to our knowledge, the optical properties of this sample are less studied. In this context, spectroscopic ellipsometry (SE), known to be a very useful, non-destructive, and accurate manner technique for investigating optical properties, is a state-of-the-art characterization technique used to analyze the optical properties and thickness of dielectric material.
In this paper, we prepared La1.4(Sr0.95Ca0.5)1.6Mn2O7 sample by a modified sol–gel method.
We have investigated the structural and the optical properties in the wavelength range of 300–800 nm by spectroscopic ellipsometry.
2 Theoretical
Spectroscopic ellipsometry is a sensitive measurement and powerful polarization-dependent technique that uses polarized light to characterize the material. It deals with the relative changes in the amplitude and the phase of a linearly polarized incident light when it interacts with a sample. It is particularly suited for the structural analysis of dielectric materials delivering information on optical response, layer thickness, homogeneity, and surface roughness, as well as orientation, anisotropy, and crystallinity of both bulk materials [17–20].
An ellipsometric system measures ellipsometric angles (ψ and Δ), where ψ gives the ratio between the absolute values of these two electric field components and Δ is the phase difference between them. These angles record the changes in polarization state of the incident light upon reflection, and they are related to the optical and structural properties of the samples. They are determined using the following formula:
where the quantities R p and R S are the complex Fresnel’s reflection coefficient of the light polarized parallel (R p) and perpendicular (R S) to the plane of incidence, respectively.
Ellipsometery is primarily used to obtain optical constants n and k, where n is the refractive index and k is the extinction coefficient, described as follows: \(\tilde {{n}}\left (\lambda \right )=n\left (\lambda \right )+ik\left (\lambda \right )\)
The complex dielectric function \(\tilde {{\varepsilon } }\left (\lambda \right )=\left ({\varepsilon _{1} \left (\lambda \right )+i\varepsilon _{2} \left (\lambda \right )} \right )\) as well as n and k is defined by
where ϕ is the angle of incidence.
3 Experiment
The microstructures of ceramic materials, in general, and CMR materials, in particular, are highly affected by preparation routes and heat treatments. Sol–gel routes are known to produce very high-quality, homogenous, and fine particle materials [21]. Bulk polycrystalline samples with the compositional formula La1.4(Sr0.95Ca0.05)1.6Mn2O7 were prepared by the sol–gel process. In this method, the metal nitrates taken in a stoichiometric ratio: La (NO3)3⋅6H2O, Sr (NO3)2, Mn (NO3)2⋅4H2O, and Ca (NO3)2⋅4H2O, were dissolved in an aqueous solution. Citric acid was added in a 1: 1 ratio and the pH was adjusted to a value between 6.5 and 7.0 by adding ammonia. After obtaining a sol on slow evaporation, a gelating reagent ethylene glycol was added and heated between 165 and 170∘C to obtain a gel. On further heating, this solution yielded a dry fluffy porous mass (precursor), which was calcined at 700∘C for 5 h. Then, the powder was pressed into circular pellets. These pellets were sintered at 900∘C in air for 10 h.
The phase purity of the sample was confirmed by X-ray diffraction (Siemens D5000 X-ray diffractometrer, with monochromatized Cu K α radiation = 1.5406 Å). The XRD data were also used for refining the lattice parameters by means of Rietveld analysis [22] (using FULL0020PROF program software).
Spectroscopic ellipsometry measurements on the sample were performed using an automatic ellipsometer, SOPRA GES 5E. The system uses a 75-W xenon lamp (185–2000 nm), a rotating polarizer, an auto tracking analyzer, a double monochromator, and a single-photon-counting photomultiplier detector. The wavelength region was 300–800 nm (1.4–4 eV) and the incident angle was 70∘ for measurements. Most surfaces are not perfectly smooth at microscopic level. All measurements were performed at room temperature.
4 Results and Discussions
4.1 X-ray Analysis
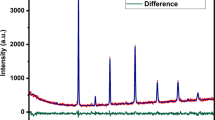
Figure 1 exhibits X-ray-diffraction patterns at room temperature. The observed and calculated profiles as well as their difference are plotted. The sample La1.4(Sr0.95Ca0.05)1.6Mn2O7 is single phase without a detectable secondary phase, within the sensitivity limits of the experimental (a few percent). The diffraction peaks are indexed with respect to the tetragonal perovskite-type structure with a space group of I4/mmm. The quality of the agreement is evaluated through the adequacy of the fit indicator χ 2. From the Rietveld refinement, we determined the lattice constants of the sample under investigation. So, the fitting results show that the lattice parameters of the compound are a = 3.8714 Å and c = 19.3267 Å (Table 1). The structure parameter values obtained for this sample are in good agreement with those reported in the literature [16].

Rietveld refinement of XRD data for La1.4(Sr0.95Ca0.05)1.6Mn2O7. The vertical bars show the Bragg reflections. The difference in the pattern between the data and the calculation is shown at the bottom of the figure
A presentation of the structure La1.4(Sr0.95Ca0.05)1.6Mn2O7 is shown in Fig. 2. Two infinite MnO6 sheets are connected in the c direction between the rock-salt layers.

The crystal structure of La1.4(Sr0.95Ca0.05)1.6Mn2O7 projected along the c-axis. The MnO6 octahedra are shaded and the (La, Sr, Ca) sites are shown as spheres. P and R refer to the 12- and 9-coordinate cations, respectively. The unit cell is shown with a solid line
The deviation from the ideal structure of a given perovskite can be estimated from the tolerance factor (t) introduced by Goldschmidt [23, 24], calculated by
In general, the perovskite structure is stable in the region 0.75 < t < 1.06, and the symmetry is as high as the t value is close to 1. This parameter is an indication of how far the atoms can move from the ideal packing and still belong to the perovskite structure. We deduce that the tolerance factor is t = 1.018 and so the structure is stable.
We have calculated the average crystallite size by using Scherer’s formula, Williamson–Hall, and SEM. Scherer’s formula can be written as [25]
where constant k depends upon the shape of the crystallite size (= 0.89, assuming the circular grain), β is the full width at half maximum (FWHM) of intensity vs. 2 θ profile, λ is the wavelength of the Cu K α radiation (0.15406 nm), and θ is the Bragg diffraction angle of the most intense peak (110). The value is listed in Table 2.
The average crystallite size values have been estimated from the full width at half maximum of X-ray diffraction peaks. The effects of synthesis and instrumental and processing conditions have been taken with care while making the calculation of crystallite sizes. The broadening of Bragg reflections due to micro strains was considered to have angular dependence and is given by β strain = 4εtanθ where β strain is the peak shift due to the strain, \(\varepsilon =\left ({\frac {\Delta d}{d}} \right )\) is a coefficient related to the strain, and θ is the Bragg angle. The micro strains include the effects of structural defects such as dislocations, stacking faults, twin boundaries, and intergrowths. The dependence of the size effect is given by the Scherer formula: \(\beta _{\text {size}} =\frac {k\lambda } {D_{\text {W-H}} \cos \theta }\), where k is the grain shape factor (for a spherical grain, k = 0.89), λ is the wavelength of Cu K α radiation (λ = 1.5406 Å), and D W−H is the thickness of the crystal. In the present investigation, only the prominent peaks have been considered. The instrument broadening effect has been eliminated by subtracting the full width at half maximum (β 0) values from β size at respective Bragg peaks of a standard sample. Finally, the complete expression for the full width at half maximum (FWHM) of the X-ray diffraction peaks is given by \(\beta _{hkl} =\frac {k\lambda }{D_{\mathrm {W}-\mathrm {H}} \cos \theta } +4\varepsilon \tan \theta \). A plot between β h k l cosθ and sin θ (Fig. 3) gives a straight line, and from the values of intercept on the y-axis, the average crystal sizes have been calculated and are found to be 8 nm (Table 2).

The W–H analysis plots of the La1.4(Sr0.95Ca0.05)1.6Mn2O7 sample. The inset shows typical scanning electron micrography (SEM)
It is worth remarking that the particle size, calculated in the present system using the Williamson–Hall technique, is larger than that obtained from the Debye–Schere technique because of the broadening of peaks due to the presence of the strain in the W–H technique.
One may observe from the inset of Fig. 3 (SEM micrograph) that the grain size (D SEM) and surface morphology are reasonably homogenous, uniform and dense. We can consider that the particle size obtained by SEM is larger than those calculated by XRD (Table 2). This can be indicating that each particle observed by SEM is formed by several grains crystallized [26].
4.2 Optical Characterization
The ellipsometric angles tan ψ and cos Δ are shown in Fig. 4. Both spectra exhibit oscillations over the entire wavelength range.

ψ and Δ spectra as function of wavelength at the incident angle of 70∘ for the sample La1.4 (Sr0.95Ca0.05)1.6Mn2O7
When an electromagnetic wave falling on a sample, a part of the wave is absorbed through the surface and another part is reflected. As the wave interacts with the sample, the electron orbits within the particle’s constituent molecules are perturbed periodically with the same frequency as the electric field of the incident wave. Thus oscillations observed in the spectra of tan ψ and cos Δ are on the basis of the perturbation of electrons.
Under the influence of the strong relation between the electronic polarizability of ions, the local field inside materials, and the optical constants, this latter play an important role in optical communication and designing of the optical devices.
Figure 5 shows the dependence of refractive index n and extinction coefficient k with the incident wavelength for the sample, that were derived from spectroscopic ellipsometry measurements, with an angle of incidence of 70∘.

The refractive index and extinction coefficient k in the wavelength range of 300–800 nm for La1.4(Sr0.95Ca0.05)1.6Mn2O7
The n– λ curves exhibit three important features. On the one hand, this plot has a maximum index at λ = 450 nm which can be attributed to the spin-glass-like (SG-like) transition induced from competing ferromagnetic (FM) double exchange (DE) and antiferromagnetic (AFM) super exchange (SE) interactions [27]. On the other hand, the second peak position of the refractive index is around 590 nm (2.1 eV) and, above this value, the index n subsequently falls rapidly, showing dispersion with wavelength. The peak in the refractive index may be arising from the direct band gap transition and this dispersion for the sample indicates sharp increase at higher photon energy corresponding to fundamental absorption of energy across the bandgap and the minimum observed in a wavelength λ≈530 nm correspond an insulator to metal transition. These curves are comparable with those observed in the literature [28].
From the spectral dependence of the extinction coefficient, we can see the same variation as the refractive index except that the values of k are low which indicates very low optical losses due to absorption. At higher photon energy, k undergoes an important increase which is due to the fundamental absorption across the band gap.
The extinction coefficient can be related to the absorption coefficient α using the relation: \(\alpha =\frac {4\pi k}{\lambda } \), where λ is the wavelength of the incident photon. The spectral behavior of the absorption coefficient in 300–800 nm is shown in Fig. 6a. The absorption coefficient is high for the wavelength below 400 nm. It decreases at first with increasing wavelength indicating a transition insulating metal about λ = 530 nm, and then, around 2.1 eV (≈590 nm), the onset of the optical absorption is observed.

a The absorption coefficient α, b plot of (α h υ)1/2, c variation of (αhυ)2 as a function of photon energy (h υ) for the La1.4(Sr0.95Ca0.05)1.6Mn2O7 sample
The optical energy gap (E g) was analyzed by the empirical equation according to the value of m as follows: (α h υ)m = A(h υ−E g ) [29, 30], where A is constant, E g is the optical energy gap, h is Planck’s constant, υ is the frequency of the incident radiation, and m determines the type of the optical transition. m = 2 for direct allowed transition, and for indirect allowed transition, the value of m equals 0.5.
The indirect energy gap was determined by plotted (α h υ)0.5 as a function of photon energy (Fig. 6b). From this figure, we can observe two clear slopes as was expected for a direct gap material [31].
Figure 6c displays the plots of (α h υ)2 vs. photon energy (h υ) for direct allowed transition to determine the band gap of the sample. The value of the energy gap E g is obtained by extrapolating the linear portion to meet the energy axis (i.e., where (α h υ)2 = 0). The calculated optical band gap energy is 2.25 eV. This means that the absorption edge is 550 nm.
The imaginary and real parts of the dielectric constant can be estimated from the following relations:
The dependence of real and imaginary parts of the dielectric function with wavelength, derived from ellipsometry data, is presented in Fig. 7. The imaginary part is related to the electronic band structure and describes the absorptive behavior while the real part describes the electronic polarizability of the double perovskites. According to Fig. 7, we can notice that ε 1 and ε 2 have the same looks as n and k respectively.

Variation of real \(\mathcal {E}_{1}\) and imaginary \(\mathcal {E}_{2}\) parts of the dielectric functions vs. wavelength for the La1.4(Sr0.95Ca0.05)1.6Mn2O7 sample
Another important two parameters which can be used to describe the optical transitions for electrons in this investigated material were the surface energy loss function (SELF), which describes the electron transitions in thin material and can be determined by the relation \(\text {SEL}=\frac {\varepsilon _{2}} {(\varepsilon _{1} +1)^{2}+\varepsilon _{2}^{2}} \) [32], and the volume energy loss function (VELF), which describes the electron transitions in the bulk materials and can be derived from the relation \(\text {VEL}=\frac {\varepsilon _{2}} {{\varepsilon _{1}^{2}} +{\varepsilon _{2}^{2}}} \) [33], which the value of SELF/VELF describes the electron transitions in this material at low and high energy [33]. The curves of SELF, VELF, and SELF/VELF were plotted as functions of photon energy as shown in Fig. 8.

a Surface energy loss, b volume energy loss, c SEL/VEL as a function of photon energy
From this figure, two transitions were seen: one is the insulator–metal transition and the second is the spin-glass-like (SG-like) transition. It was confirmed with the previous results of spectra n and k.
The refractive index can be fitted to the Wemple–DiDomenico (WDD) dispersion relation that is a single-oscillator model [33].
where E 0 is the single-oscillator energy, which can directly correlate with the optical energy gap by an empirical formula, while E d is the dispersion energy, which relates to the average strength of interband optical transitions. By plotting (n 2−1)−1 vs. (h υ)2 and fitting the relation to a straight lines (Fig. 9), E 0 and E d can be determined from the slope (E 0 E d)−1 and incident photon energy E = 0 intercept (E 0/E d).

Plots of (n 2−1)−1 vs. (hυ)2 are used to determine E 0 and E d
Values of E 0 and E d equal 2.525 eV and 16.7 meV respectively.
On the other hand, the single-oscillator model parameters E 0 and E d are connected to M−1 and M−3 moments of the imaginary part of the complex dielectric constant ε 2(hυ) optical spectrum, by the following relation:
The values of M−1 and M−3 are 6.62 10−4 and 1.04 10−4 eV−2.
The optical conductivity is considered as a powerful probe in the determination of the electronic properties of a variety of materials. Generally, when a system is submitted to an external electric field, a redistribution of charges occurs and the current is induced.
The optical conductivity is related to imaginary parts complex constant ε 2 by the relation:
where ω = 2π υ is the angular frequency.
In Fig. 10, the optical conductivity of the sample is plotted against the photon energy and was in good agreement with the reported in the literature [34].

Photon energy dependence of the optical conductivity for the La1.4(Sr0.95Ca0.05)1.6Mn2O7 sample
In physics, the dissipation factor tanδ is a measure of the loss-rate of power of a mechanical mode, such as an oscillation, in a dissipative system. It can be calculated according to the following equation [35, 36]:
The variation of tanδ for the investigated material with h υ is shown in Fig. 11. We can see the same behavior observed in the spectra of extinction coefficient k.

Variation of the dissipation factor \(\tan \delta \) vs. photon energy
5 Conclusion
In conclusion, La1.4(Sr0.95Ca0.05)1.6Mn2O7 was prepared by the sol–gel method. The structural, morphology surface and optical properties have been studied. The XRD pattern reveals that the sample has a single-phase perovskite structure with I4/mmm space group. The optical constants (n, k) and band gaps (E g) were determined by spectroscopic ellipsometry in the 1.4–4 eV photon energy range. The results suggest that the transmission ellipsometric parameters ψ and Δ reveal oscillations.
The refractive index dispersion parameters were determined using the single oscillator model proposed by Wemple-DiDomenico (WDD).
The conductivity vs. photon energy plots indicate:
-
The coexistence of AFM and FM interactions
-
The insulator–metal transition (I–M)
The E g value, obtained by analyzing with the (α h υ)2 vs. photon energy curve, was estimated to be 2.25 eV.
References
Gupta, A.K., Bhalla, G.L., Neeraj, K.: Magnetic phase diagram of double-layered La2−2x Ca1+2x Mn2O7 manganite. J. Phys. Chem. Solids 67, 2358–2364 (2006)
Han, L.A., Chen, C.: Magnetocaloric and colossal magnetoresistance effect in layered perovskite La1.4Sr1.6Mn2O7. J. Mater. Sci. Technol. 26, 234–236 (2010)
Ehsani, M.H., Ghazi, M.E., Kameli, P.: Effects pf pH and sintering temperature on the synthesis and electrical properties of the bilayered LaSr2Mn2O7 manganite prepared by the sol-gel process. J. Mater. Sci. 47, 5815–5822 (2012)
Moritomo, Y., Asamitsu, A., Kuwahara, H., Tokura, Y.: Giant magnetoresistance of manganese oxides with a layered perovskite structure. Nature 380, 141–144 (1996)
Salamon, M.B., Jaime, M.: The physics of manganites: structure and transport. Rev. Mod. Phys. 73, 583– 628 (2001)
Dagotto, E., Hotta, T., Moreo, A.: Colossal magnetoresistant materials: the key role of phase separation. Phys. Rep. 344, 1–153 (2001)
Sreedhar, K., McElfresh, M., Perry, D., Kim, D., Metcalf, P., Honig, J.M.: Low-temperature electronic properties of the Lan + 1NinO3n + 1(n =2,3, and ∞) system: evidence for a crossover from fluctuating-valence to Fermi-liquid-like. J. Solid State Chem. 110, 208–215 (1994)
Zhang, Z., Greenblatt, M., Goodenough, J.B.: Synthesis, structure, and properties of the layered perovskite La3Ni2O7−δ . J. Solid State Chem. 108, 402–409 (1994)
Rao, C.N.R., Ganguly, P., Singh, K.K., Mohan Ram, R.A.: A comparative study of the magnetic and electrical properties of perovskite oxides and the corresponding two-dimensional oxides of K2NiF4 structure. J. Solid State Chem. 72, 14–23 (1988)
Sharma, I.B., Singh, D., Magotra, S.K.: Effect of substitution of magnetic rare earths for La on the structure, electric transport and magnetic properties of La2SrFe2O7. J. Alloys Compd. 269, 13–16 (1998)
Kimura, T., Tokura, Y.: Layered magnetic manganites. Annu. Rev. Mater. Sci. 30, 451–474 (2000)
Chen, C.C., Shen, C.H., Liu, R.S., Lin, J.G., Hoang, C.Y.: Synthesis and characterization of the colossal magnetoresistance manganite La1.2(Sr1.4Ca0.4)Mn2O7 by citrate gel. Mater. Res. Bull. 37, 235–246 (2002)
Liu, J.W., Chen, G., Li, Z.H., Lu, Z., Zhang, Z.G.: Synthesis and colossal magnetoresistance effect of layered perovskites Sm2−2xSr1+2xMn2O7 (x = 0.2,0.4,0.5). Mater. Chem. Phys. 105, 185–188 (2007)
Dudric, R., Goga, F., Neumann, M., Mican, S., Tetean, R.: Magnetic properties and magnetocaloric effect in La1.4−xCexCa1.6Mn2O7 perovskites synthesized by sol-gel method. J. Mater. Sci. 47, 3125–3130 (2012)
Zhao, X., Chen, W., Zong, Y., Diao, S.L., Yan, X.J., Zhu, M.G.: Structure, magnetic and magnetiocaloric properties in La1.4Sr1.6−xCaxMn2O7 perovskite compounds. J. Alloys Compd. 469, 61–65 (2009)
Shen, C.H., Liu, R.S., Hu, S.F., Lin, J.G., Huang, C.Y., Sheu, H.S.: Structural, electrical and magnetic properties of two-dimensional La1.2(Sr1.8−xCax)Mn2O7 manganites. J. Appl. Phys. 86, 2178–2184 (1999)
Lautenschlager, P., Garriga, M., Logothetidis, S., Cardona, M.: Interband critical points of GaAs nd their temperature dependence. Phys. Rev. B 35, 9174–9189 (1987)
Yang, P., Guo, M., Shi, M., Meng, X., Huang, Z., Chu, J.: Spectroscopic ellipsometry of SrBi2Ta2−xNbxO9 ferroelectric thin films. J. Appl. Phys. 97, 106106–106106-3 (2005)
Yu, J., Huang, Z.M., Meng, X.J., Sun, J.L., Chu, J.H., Tang, D.Y.: Infrared optical properties of LaNiO3-platinized silicon and PbZrxTi1−xO3-LaNiO3-platinized silicon heterostructures. Appl. Phys. Lett. 78, 793 (2001)
Moret, M.P., Devillers, M.A.C., Worhoff, K., Larsen, P.K.: Optical properties of PbTiO3, PbZrxTi1−xO3, and PbZrO3 films deposited by metalorganic chemical vapor on SrTiO3. J. Appl. Phys. 92, 468–474 (2002)
Lisboa-Filho, P.N., Mombru, A.W., Pardo, H., Leite, E.R., Ortiz, W.A.: Extrinsic properties of colossal magnetoresistive samples. Soli. State Commun. 130, 31–36 (2004)
Rietveld, H.M.: A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 2, 65–71 (1969)
Goldschmidt, V.M.: Skrifter Norske Videnskaps-Akad. Oslo I. Mater. Natur. 2, 7 (1926)
Ganguly, P., Rao, C.N.R.: Crystal chemistry and magnetic properties of layered metal oxides possessing the K2NiF4 or related structures. J. Solid State Chem. 53, 193–216 (1984)
Taylor, A.: X-ray Metallography. Wiley, New York (1961)
Gutiérrez, J., Pena, A., Barandiaran, J.M., Pizarro, J.L., Hernandez, T, Lezama, L., Insausti, M., Rojo, T.: Structural and magnetic properties of La0.7Pb0.3(Mn1-xFex)O3(0 ≤x≤0.3) giant magnetoresistance perovskites. Phys. Rev. B 61, 9028–9035 (2000)
Chatterjee, S., Chou, P.H., Chang, C.F., Hong, I.P., Yang, H.D.: Lattice effects on the transport properties of (R, Sr)3Mn2O7(R =La,Eu, and Pr). Phys. Rev. B 61, 6106–6113 (2000)
Sdiri, N., Boukhachem, A., Dhahri, E.: Optical investigations of La0.7Ca0.3−x K xMnO3(x =0.00, 0.05 and 0.10) probed by spectroscopic ellipsometry. Ceramics-Silikaty 56, 95–101 (2012)
Liu, H.L., Yoon, S., Cooper, S.L., Cheong, S.W., Han, P.D., Payne, D.A.: Probing anisotropic magnetotransport in manganese perovskites using Raman spectroscopy. Phys. Rev. B 58, 115–118 (1998)
Tauc, J.C.: Optical Properties of Solids. North-Holland, Amsterdam (1972)
Pankov, J.I.: Optical Processes in Semiconductors, p 37. Dover, New York (1975)
Tan, G.L., DeNoyer, L.K., French, R.H., Guittet, M.J, Gautier-Soyer: J. Electr. Spectros. Relat. Phenome. 142, 97–103 (2004)
Wemple, S.H., DiDomenico, M.: Behavior of the electronic dielectric constant in covalent and ionic materials. J. Phys. Rev. B 3, 1338–1351 (1971)
Gan’shina, E., Loshkareva, N., Sukhorukov, Yu., Mostovshchikova, E., Vinogradov, A., Nomerovannaya, L.: Optical and magneto-optical spectroscopy of manganites. J. Magn. Magn. Mater. 300, 62–66 (2006)
Fadel, M., Fayek, S.A., Abou-Helal, M.O., Ibrahim, M.M., Shakra, A.M.: Structural and optical properties of SeGe and SeGex(x =Ln,S band Bi) amorphous films. J. Alloys Compd. 485, 604–609 (2009)
Abdel-Aziz, M.M., El-Metwally, E.G., Fadel, M., Labib, H.H., Afifi, M.A.: Optical properties of amorphous GeSeTi system films. Thin Solid Films 386, 99 (2001)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Belkahla, A., Cherif, K., Dhahri, J. et al. Structural and Optical Properties of the Ruddlesden–Popper La1.4(Sr0.95Ca0.05)1.6Mn2O7 Sample Prepared by a Sol–Gel Method. J Supercond Nov Magn 29, 19–27 (2016). https://doi.org/10.1007/s10948-015-3267-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-015-3267-3