
Abstract
Superconducting Bi1.6Pb0.4Sr2Ca2Cu3O7 was prepared by a solid-state reaction using high-energy milling for the starting materials. The effects of the sintering temperature and time on the phase evolution, microstructure, and superconducting properties were examined by thermogravimetric analysis, X-ray diffraction, scanning electron microscopy, and four-point probe resistivity measurements. High-energy milling decreased the precursor particle size to approximately 30 nm with a relatively uniform distribution of multi-compositions. Because of the mechanochemical activation for the starting materials, the reaction temperature was decreased to 750 ∘C due to the decreased diffusion length required for the solid-state reaction. Bi1.6Pb0.4Sr2Ca2Cu3O7 prepared by high-energy milling showed good superconducting properties with T c∼ 100 K, which is superior to the same material prepared by ball milling and sol-gel routes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Since the discovery of the superconducting Bi-Sr-Ca-Cu-O (BSCCO) system by Maeda et al., there have been considerable efforts aimed at improving its physical and electrical properties [1]. BSCCO has three different phases based on the number of Cu-O planes (n= 1, 2, and 3), which are commonly referred to as 2201, 2212, and 2223 phases with critical temperatures (T c) of approximately 20, 80, and 110 K, respectively. Among these, the most promising phase for practical applications is the 2223 phase because of its higher critical temperature. On the other hand, achieving a single 2223 phase is very difficult because of its narrow temperature stability.
Superconducting materials, including BSCCO, are commonly synthesized via a solid-state reaction through the mixing and calcination of oxide or carbonate starting materials. This method, however, has several disadvantages, such as the nonuniform distribution of starting materials, significant impurity content and long processing time; the synthesis of a high-quality superconductor oxide needs to overcome all these barriers. Therefore, several alternative methods have been proposed to obtain an improved superconducting powder with a stoichiometric composition, including the utilization of recently introduced equipment and wet-processing techniques [2–4]. The use of planetary milling and attrition milling under dry conditions results in high-quality superconducting oxide powders [5–7]. On the other hand, fluid energy comminuting was also reported to be effective in decreasing the particle size of the starting materials as well as increasing the critical current (I c) [4]. Despite these efforts, however, the synthesis of BSCCO powder requires a long heat treatment time, 840–860 ∘C for up to several days, which needs to be reduced.
With this background, this study examined the effects of high energy milling for the synthesis of high quality BSCCO, particularly to decrease the processing time. Although high-energy milling has been used for ceramic processing in many cases [8], to the best of authors’ knowledge, there are no reports on the use of high-energy milling for the synthesis of BSCCO superconducting materials. Therefore, this study examined the effects of high-energy milling on the solid-state synthesis of BSCCO. A (Bi, Pb)-Sr-Ca-Cu-O precursor powder was high-energy-milled using very fine ZrO2 grinding media to decrease the heat treatment temperature and time by mechanochemical activation of the starting materials. For comparison, BSCCO powder was also synthesized via a solid-state reaction using two different milling methods, ball milling and shaker milling. At the same time, wet-processing using a sol-gel route was also applied for the synthesis of BSCCO.
2 Experimental
Bi2O3, PbO, SrCO3, CaCO3, and CuO (all from Sigma-Aldrich Chemicals with a purity >99.9 %) at a molar ratio of 1.6:0.4:2.0:2.0:3.0 were used as starting materials for the synthesis of Bi1.6Pb0.4Sr2Ca2Cu3O7. The powder mixture was high-energy-milled (MiniCer, Netzsch, Germany) for 3 h using 0.45-mm ZrO2 beads at a rotor speed of 3000 rpm after adding 2 wt % of ammonium polycarboxylate dispersant (Cerasperse 5468-CF, San Nopco, Korea) in ethanol.
For the sol-gel method, Bi1.6Pb0.4Sr2Ca2Cu3O7 was prepared using their corresponding high-purity metal nitrates as the starting materials. After dissolving citric acid in distilled water, all metal nitrates weighed at a stoichiometric ratio were added into the citric acid solution followed by the addition of ethylene glycol. The viscous solution was heat-treated at 300 ∘C to obtain a precursor powder. More details on the sol-gel citrate route can be found elsewhere [9]. At the same time, mixed powders with the same stoichiometry were also prepared by 24 h of ball milling and 3 h of shaker milling (mini milling). The milled powder was dried completely using a rotary evaporator at 90 ∘C followed by grinding and sieving. The precursor powder was then placed into an alumina crucible and heat-treated in a box furnace for various temperatures (500–800 ∘C) at a heating rate of 5 ∘C/min for 6 h. The calcined powder was ground using an agate mortar and pressed uniaxially at 500 MPa. The pellets, 1 cm in diameter, were sintered at 820, 840, and 860 ∘C for 24, 72, and 120 h at a heating rate of 5 ∘C/min to produce the BSCCO superconducting phase.
Thermogravimetric and differential thermal analysis (TG-DTA, SDT Q600 TA Instrument) of the highenergy-milled mixture were performed to characterize the thermal decomposition of the precursor powder. The phases generated at each calcination step and sintering were confirmed by X-ray diffraction (XRD: X’Pert-PRO MPD, PANalytical using the Cu K α line, 40 kV and 30 mA) over the range, 10 ∘ –90 ∘ 2 𝜃. The morphology of the particles and sintered bodies were observed by scanning electron microscopy (SEM: S-4800, Hitachi at 15 kV and 10 μA). The mean particle size was estimated from the SEM images by measuring the maximum and minimum diameters of 100 particles using image analysis software (SigmaScan, Systat Software, USA).
The superconducting properties of the sintered pellets were measured using the four-point probe method using a cryogenic magnet system (Teslatron PT Cryogen Free, Oxford Instrument) in the temperature range, 10–300 K. The critical temperature (T c), where a material loses its resistivity and becomes a superconductor; T c (zero), the temperature, where the material begins to lose its resistivity and (T c onset), the starting point for the transition to a superconducting state. T cis defined as the temperature where it shows the maximum derivative from the resistivity vs. temperature curve and the transition width, Δ T, is determined from the initial T c(zero).
3 Results and Discussion
3.1 Characterization of the Calcined Powder
The effects of high-energy milling on the particle size of the starting material and its distribution were compared with the precursor powders prepared by the ball milling, shaker milling, and sol-gel processes. Figure 1 presents SEM images of the precursor powders after each treatment. High-energy milling resulted in a much finer starting material size than the ball milling, shaker milling, and sol-gel routes. Figure 2 shows the particle size distribution of the powders produced by high-energy milling and the other methods. The mean particle size of the precursor powder produced by high-energy milling was 28.88 nm with a narrow size distribution (15–50 nm). Figures 1 and 2 show that much smaller particle sizes in the nanometer range with a narrow distribution can be obtained by high-energy milling compared to the other processing routes. Very fine starting materials with a homogeneous distribution is desirable for enhancing the reaction rate between the constituent particles. Therefore, the powder prepared by high-energy milling was chosen for further evaluation, including the phase evolution and microstructural changes, after exposure to different temperatures.

SEM images of the precursor powder produced by a high-energy milling, b sol-gel method, c ball milling, and d shaker milling

Particle size distribution of the BSCCO precursor powder prepared by various routes
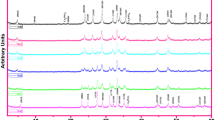
Powder XRD was performed on the high-energy-milled powder to examine the structural changes that occurred after calcination at different temperatures. Figure 3 shows SEM images of the precursor powders after calcination at 350, 550, 750, and 800 ∘C for 6 h in air, and Fig. 4 shows the corresponding XRD patterns. The precursor powder began to change its morphology from 550 ∘C, showing significant growth at 750 ∘C, according to Fig. 3. From Fig. 4, it was evident that the as-prepared amorphous powder shows only broad diffraction peaks corresponding to the starting precursor powders (Bi2O3, PbO, SrCO3, CaCO3, and CuO), indicating that no chemical reaction had occurred between the constituent phases during the milling process. After calcination at 350 ∘C, the peaks corresponding to the Bi2CuO4 phase began to appear. Further increases in the calcination temperature to 550 ∘C resulted in a significant increase in the intensity of the Bi2CuO4 peaks, while peaks corresponding to the Ca2PbO4 and Sr0.6Ca0.4CuO2 phases can also be seen. When the temperature was increased to 750 ∘C, peaks corresponding to the Bi1.6Pb0.4Sr2Ca2Cu3O7 2201 and 2212 phases were observed, whereas the peaks for Bi2CuO4 had disappeared. On the other hand, the intensity of the peaks for Ca2PbO4 also increased at this temperature. Finally, when the calcination temperature was increased further to 800 ∘C, the dominant phase was 2212 along with small traces of 2201, CuO, and Ca2PbO4. The presence of a CuO phase beyond 750 ∘C might be due to the decomposition of Bi2CuO4. These results are consistent with those reported earlier using a different preparation method [10].

SEM images of the high-energy-milled precursor powders calcined at a 350, b 550, c 750, and d 800 ∘C for 6 h

X-ray diffraction pattern of the high-energy-milled precursor powder calcined at different temperatures
Several researchers have examined the various phase transformations occurring during the synthesis of BSCCO-based superconducting materials [11–14]. Most of the XRD diffraction peaks could be indexed according to the phases formed in the ternary or quaternary systems, such as Bi-Sr-Ca, Sr-Ca-Cu, Bi-Ca-Cu, Bi-Sr-Cu, and Bi-Sr-Ca-Cu-O, as reported by Paz-Pujalt [15]. The reactions involved in the formation of Bi2Sr2Ca n Cu1+n O6+2n can be summarized as follows:
Figure 5 presents the TG-DTA results for the high-energy-milled powder. The figure shows that the total weight loss of the precursor was almost 20 %, where the weight loss beyond 750 ∘C was quite small (∼ 1.7 %). This suggests that at temperatures <750 ∘C, a solid-state reaction between the constituent particles occurred to form the BSCCO compound, which is consistent with the XRD patterns in Fig. 4. Although not shown here, the corresponding reaction completion temperature for ball milling and sol-gel route based on the TG-DTA data were 842 and 821 ∘C, respectively, which is much higher than that of the high-energy-milled sample. The lower reaction temperature for the high-energy-milled sample can be attributed to the finer particle size and uniform distribution, as shown in Figs. 1 and 2, which decreases the diffusion path required for the solid-state reaction. Figure 5 shows that there are four distinct weight loss ranges, 40–200, 200–400, 400–750, and 750–900 ∘C. The weight loss at 40–200 ∘C might be due to the removal of water adsorbed on the surface of powder, which is indicated by the endothermic peak at approximately 104 ∘C. Over the temperature range, 200–400 ∘C, the sample exhibited another endothermic peak at approximately 256 ∘C, which might be due to the elimination of the organic dispersant. An endothermic peak at 423.4 ∘C and exothermic peak at 620 ∘C along with a significant weight loss of 13.4 % was observed at 400–750 ∘C, which might be due to the formation of Bi2CuO4 and Ca2PbO4 phases. Finally, the small DTA peaks observed at 750–900 ∘C can be attributed to the formation of Bi1.6Pb0.4Sr2Ca2Cu3O7 2201 and 2212 phases.

TGA/DTA curves of the precursor powder prepared by high-energy milling
4 Sintered Powder Characterization
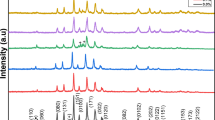
Figure 6 presents the XRD patterns of the pellets sintered at 820, 840, and 860 ∘C for 24, 72, and 120 h. The diffraction patterns correspond mainly to the 2212 and 2223 phases with orthorhombic and tetragonal structures, respectively [16–20]. This difference in crystal structures in the Cu-based superconductor oxides is due to the difference in bonding between copper and oxygen atoms [21]. The bonds between two Bi-O layers in the BSCCO system are weak due to the van der Waals force, whereas the bonds in the basal plane are much stronger [22, 23]. The insets show the low angle diffraction patterns to clearly identify the peaks corresponding to the 2212 and 2223 phases. The intensity of the 2223 phase increased with increasing sintering temperature from 820 to 860 ∘C and with increasing sintering time from 24 to 120 h at 840 ∘C. Quantitative analysis was performed based on the summation of the integrated peak intensities to estimate the volume fraction of the 2223 and 2212 phases. For example, the volume fraction of the 2223 phase was calculated using the following equation, and is summarized in Table 1.

X-Ray diffraction patterns of high-energy-milled sample sintered at different temperatures for different times in air
From Table 1, it appears that the sintering time is the predominant factor for the formation of the 2223 phase compared the sintering temperature. The decreased volume fraction of 2223 at 860 ∘C might be due to its instability at higher temperatures. Sintering at high temperatures is responsible for the oxygen loss of the sample, resulting in the formation of 2212 and 2201 phases [24]. Therefore, the optimal sintering temperature to obtain a high 2223 phase content is 840 ∘C. Matsushita et al. reported that the formation of high- T c superconducting BSCCO ceramics (T c ∼110 K for Pb = 0.2 ) was enhanced remarkably by annealing the sample at the endothermic region [25]. The formation of a 2223 phase appears to be affected by many factors, such as the sintering temperature, time, oxygen partial pressure, and composition. Because a longer time is desirable for increasing the formation of the 2223 phase, the sintering time was increased from 24 to 120 h, which resulted in a significant increase in the 2223 volume fraction, as shown in Fig. 6 and Table 1.
Figure 7 shows SEM images of the fractured samples sintered at different conditions. Plate-like grains were formed, whereas the grain size increased from 0.15 to 10 µm in diameter with increasing sintering temperature and time. The mean platelet sizes of the samples sintered at 820, 840, and 860 ∘C for 24 h were 0.15, 0.30, and 1.50 μm, respectively. This plate-like morphology is typical for BSCCO superconducting oxides, which originated from their anisotropic nature [26]. Based on Fig. 7, it appears that the size of the platelets increases with increasing sintering time.

SEM images of the high energy-milled samples sintered at a 820, b 840, and c 860 ∘C for 24 h in air; sintered at 840 ∘C for d 72 and e 120 h
5 Superconducting Properties
The different microstructures and phase contents depending on the sintering conditions also affect the electrical properties, as shown in Fig. 8, which presents the resistivity vs. temperature from 10 to 300 K. All the samples exhibited metallic behavior in a normal state (up to T c onset). In addition, all the samples showed a very narrow width of transition temperatures, indicating a single superconducting transition, except for the sample sintered at 860 ∘C. Figure 9 presents the derivatives of the resistivity (d ρ/dT) for all samples to obtain clear information on the variations of the critical temperatures according to the sintering temperatures and times. Figures 8 and 9 show that the transition temperatures are crucially dependent upon the sintering temperature and sintering time. Moreover, the T Cvalues (from d ρ/dT curves) increase with increasing sintering temperature, as shown in Table 1. On the other hand, when the sintering time was increased from 24 to 72 h at 840 ∘C, T C increased from 104.8 to 107 K, and then decreased to 104 K as the sintering time was increased further to 120 h. Overall, the samples sintered at 840 ∘C for 72 h showed the best T C results. On the other hand, the relatively low T C value obtained from the samples sintered for 120 h was attributed to decomposition of the 2223 phase to the 2201 phase. These values are comparable to those reported for BSCCO superconductors [27–32].

Normalized resistivity vs. absolute temperature of BSCCO prepared by high-energy milling

Critical temperature (T c derivative) vs. temperature for the high-energy-milled sample sintered at 820, 840, and 860 ∘C for 24 h, and 840 ∘C for 72 and 120 h
6 Conclusions
This paper presented the experimental results on the solid-state synthesis of homogenous BSCCO superconductor oxide by high-energy milling for the starting materials. The phase evolution, microstructural changes in the sample, and superconducting properties have been examined. High-energy milling was found to be quite effective in decreasing the particle size of the precursor powder with a uniform distribution, which resulted in a drastic decrease in the solid-state reaction temperature up to ∼ 100 ∘C because of the reduced diffusion length required for the reaction. BSCCO superconducting materials synthesized by high-energy milling followed by calcination and sintering revealed a mixed system of 2212 and 2223. Based on the electrical measurements, the optimal sintering condition was found to be 840 ∘C with a holding time of 72 h.
References
Hiroshi, M., Tanaka, Y., Fukutomi, M., Asano, T.: Jpn. J. Appl. Phys. 27, L209–L210 (1988)
Ahn, J.H., Oh, S.: J. Alloy Comp. 504, S292–S294 (2010)
Patel, R.H., Nabialek, A., Niewczas, M.: Supercond. Sci. Technol. 18, 317–324 (2005)
Yao, Y.X., Li, Y.N., Feng, R.B., Luan, W.Z., Wang, Y.: Phys. C 386, 106–109 (2003)
Aloysius, R.P., Guruswamy, P., Syamaprasad, U.: Supercond. Sci. Technol. 18, L23–L28 (2005)
Lee, H., Park, H., Lee, D.: Metals Mater. 6, 473–489 (2000)
Kim, W.-J., Kwon, S.-C., Lee, H.J., Lee, H.-G., Hong, G.-W., Kuk, I.-H.: Phys. C 294, 147–155 (1998)
Fu, Z.-F., Liu, P., Ma, J.-L.: Mat. Sci. Eng. B 193, 32–36 (2015)
Raju, K., Yoon, D.H.: J. Supercond. Nov. Magn. 27, 1285–1292 (2014)
Aiko, A.: Jpn. J. Appl. Phys. 29, L270–L272 (1990)
Wang, M., Xiong, G., Tang, X., Hong, Z.: Phys. C 210, 413–416 (1993)
Hu, Q.Y., Liu, H.K., Dou, S.X.: Phys. C 250, 7–14 (1995)
Hatano, T., Aota, K., Ikeda, S., Nakamura, K., Ogawa, K.: Jpn. J. Appl. Phys. 27, L2055–L2058 (1988)
Grindatto, D.P., Grivel, J.-C., Grasso, G., Nissen, H.-U., Flükiger, R.: Phys. C 298, 41–48 (1998)
Paz-Pujalt, G.R.: Phys. C 166, 177–184 (1990)
Green, S.M., Mei, Y., Manzi, A.E., Luo, H.L., Ramesh, R., Thomas, G.: J. Appl. Phys. 66, 728–734 (1989)
Pandey, D., Mahesh, R., Sing, A.K., Tiwari, V.S., Kak, S.K.: Solid State Commun. 76, 655–658 (1990)
Pandey, D., Mahesh, R., Sing, A.K., Tiwari, V.S., Kak, S.K.: Solid State Commun. 75, 499–502 (1990)
Pandey, D., Sing, A.K., Mahesh, R., Tiwari, V.S., Kak, S.K.: Phys. C 173, 476–486 (1991)
Sequeira, A., Rajagopal, H., Sastry, P.V.P.S.S., Yakhmi, J.V., Iyer, R.M.: Phys. C 173, 267–273 (1991)
Crawford, M.K., Harlow, R. L., McCarron, E. M., Farneth, W. E., Axe, J. D., Chou, H., Huanget, Q.: Phys. Rev. B 44, 7749–7752 (1991)
Luo, J.S., Lee, H.G., Sinha, S.N.: J. Mater. Res. 9, 297–304 (1994)
Tsuneyuki, K., Tomoichi, K., Shin-Pei, M.: Jpn. J. Appl. Phys. 29, L412—L415 (1990)
Pissas, M., Niarchos, D., Christides, C., Anagnostou, M.: Supercond. Sci. Technol. 3, 128–133 (1990)
Matsushita, T., Suzuki, A., Teramoto, K., Okuda, M., Naito, H.: Supercond. Sci. Technol. 4, 721–724 (1991)
Tarascon, J.M., Le Page, Y., Barboux, P., Bagley, B. G., Greene, L. H., McKinnon, W. R., Hull, G. W., Giroud, M., Hwang, D. M.: Phys.Rev. B 37, 9382–9389 (1988)
Khalil, S.M.: J. Phys. Chem. Solids 62, 457–466 (2001)
Hannachi, E., Ben Salem, M.K., Slimani, Y., Hamrita, A., Zouaoui, M., Ben Azzouz, F., Ben Salem, M.: Phys. B 430, 52–57 (2013)
Ma, K., Pierre, A.C.: J. Mater. Res. 9, 286–291 (1994)
Kumar, J., Sharma, D., Ahluwalia, P.K., Awana, V.P.S.: Mater. Chem. Phys. 139, 681–688 (2013)
Kocabas, K., Gokce, M., Ciftcioglu, M., Bilgili, O.: J. Supercond. Nov. Magn. 23, 397–410 (2010)
Sotelo, A., Rasekh, Sh., Constantinescu, G., Amaveda, H., Torres, M.A., Madre, M.A., Diez, J.C.: J. Eur. Ceram. Soc. 34, 2977–2982 (2014)
Acknowledgments
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. NRF-2014K2A4A1035029). This program was also supported by the Indonesian research fund for competitive research for advanced materials and nanotechnology 2014.
Conflict of interests
The authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nono Darsono holds a PhD degree at Yeungnam University.
Agung Imaduddin holds a PhD degree at Indonesian Institute of Sciences.
Kati Raju holds a PhD degree at Yeungnam University.
Dang-Hyok Yoon holds a PhD degree at Yeungnam University.
Rights and permissions
About this article

Cite this article
Darsono, N., Imaduddin, A., Raju, K. et al. Synthesis and Characterization of Bi1.6Pb0.4Sr2Ca2Cu3O7 Superconducting Oxide by High-Energy Milling. J Supercond Nov Magn 28, 2259–2266 (2015). https://doi.org/10.1007/s10948-015-3036-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-015-3036-3