
Abstract
We report several ab initio calculations performed for Sr2CoB′O6 (B′ = Mo, Re) by means of the Density Functional Theory and the Linearized Augmented Plane Waves method with spin polarization. For the calculations, the exchange and correlation potentials were included through the local density LDA+U approximation with B3PW91. Density of states (DOS) study was carried out considering both up and down spin polarizations by the Generalized Gradient Approximation (GGA). From the Murnaghan state equation, we calculate the cell dimensions that minimize the total energy. Our results of DOS calculations show that the Sr2CoMoO6 material presents a metallic behavior, while Sr2CoReO6 reveals a half-metallic nature with conductor behavior for the spin down orientation and semiconducting feature for spin up channel. It was observed close to Fermi level that the low-energy spin down states of Co are responsible by the majority contribution to conduction band. The calculated effective cell magnetic moment of the Sr2CoReO6 compound evidences a value 2.02 μ B , which is close to an integer number as expected for a half-metallic material.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Double perovskites with formula A2BB′O6, where A represents an alkaline earth, B and B′ are metal transition magnetic and non-magnetic ions and O is the oxygen, have been known for many years [1] but recently the study of these materials has increased due to the various technological applications in the design of magnetic memories or tunnel junctions and other magnetic devices in the spintronics area [2]. Perovskite materials have attracted special attention in many applied and fundamental areas of solid-state and advanced material sciences due to the exotic magnetic properties such as colossal magnetoresistance [3], half-metallicity [4] and magnetoelectricity [5]. Complex perovskite oxides generally have the formula A2BB′O6. These oxides result from the ordering of B and B′ cations on the octahedral site of primitive perovskite unit cell. Magnetic complex perovskites A2BB′O6, where A is an alkaline earth and B, B′ are magnetic and non-magnetic transition metal, respectively, were discovered by Longo and Ward in 1960s [1]. Nevertheless, the half-metallic feature of the Sr2FeMoO6 was only established by Kobayashi et al. in 1998 [3]. This exotic property is characterized by the differentiated conducting response of spin up and spin down orientations. The density of states (DOS) as a function of energy clearly evidences that majority spin component shows a energy gap at the Fermi level, as the insulating materials, and the other spin orientation is continuous at the Fermi level, due the strong hybridization of Fe-3d(t 2g ) and O-2p states. The extensive half-metallicity studies in double perovskite materials are related with the probable technological applications in spintronic devices, such as spin valves, sources for spin polarized electrons and magnetic information storage systems. The aim of this work is to carry out a detailed ab initio theoretical study of the complex perovskites Sr2CoReO6 and Sr2CoMoO6, which were experimentally analyzed and reported as antiferromagnetic materials with low Néel temperatures of T N =65 K and T N =37 K, respectively [6, 7]. Our calculations show that Sr2CoReO6 behaves as a half-metallic with a total magnetic moment which is an integer number of Bohr magnetons. On the other hand, Sr2CoMoO6 presents a metallic behavior.
2 Calculation Method
Calculation of band and electronic structure for Sr2BB′O6 complex perovskite can be seen as a many body problem of ions and electrons. These were performed employing the FP-LAPW method, in the framework of Density Functional Theory (DFT) as implemented in the WIEN2k code [8]. The FP-LAPW consists of the calculation of solutions for the Kohn-Sham equations by first principles methods. In the calculations reported here, we use a parameter RMT∗K max=8, which determines matrix size (convergence), where K max is the plane wave cut-off and RMT is the smallest of all atomic sphere radii. We have chosen muffin-tin radii (RMT) of 2.5, 1.95, 1.95 and 1.73 for Sr, Co, Mo and O, respectively, for Sr2CoMoO6 and 2.5, 1.96, 1.96 and 1.69 for Sr, Co, Re and O, respectively, for Sr2CoReO6. The exchange and correlation effects were treated using the local density LDA+U approximation with B3PW91 and the Generalized Gradient Approximation (GGA) for the spin polarization [9]. This potential considers the difference between the electronic densities for the two distinct spin orientations. The self-consistent calculations are considered to be convergent when the total energies of two successive iterations agreed within 10−4 Ry. We adjusted the Fermi energy to zero. The integrals over the irreducible Brillouin zone are performed up to 196 k-points.
3 Results and Discussion
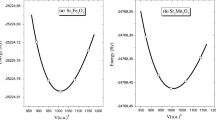
In order to obtain the most accurate results, we have determined the optimal lattice parameters corresponding to the minimal energy value. Figure 1 shows the total energy as function of c/a factor. For both materials Sr2CoReO6 and Sr2CoMoO6 total energy was calculated by fittings with the Murnaghan state equation. From the total energy as a function of volume we determine the ideal lattice parameters a and c. Calculations were performed choosing the tetragonal crystallographic structure I4/m (#87). We obtain lattice parameters a=5.533 Å, c=7.860 Å for Sr2CoMoO6 and a=5.533 Å, c=7.859 Å for Sr2CoReO6, which correspond to c/a factors of 1.42. These results are in accordance with experimental results reported in reference [6].

Total energy as a function of the c/a factor for the Sr2CoReO6 double perovskite, considering the I4/m space group. The inset shows the total energy for the Sr2CoMoO6 compound
As shown in Fig. 2a, for total DOS close to Fermi level, we observe that Sr2CoMoO6 evidences a metallic tendency for both up and down spin orientations. The up states appear from −7.70 eV up to 0.15 eV, in a continuous line through the Fermi level. Down states appear in the energy regime between −7.70 eV and −1.78 eV, followed by an intragap of 0.6 eV and a continuous band from −1.18 up to 0.2 eV. It is observed in Fig. 2b that Sr2CoReO6 presents a clear half-metallic nature: the spin up channel has a semiconducting behavior, with a gap of 0.27 eV through the Fermi level, and the spin down evidences a conductor feature. The up states are identified from −8.59 eV up to −0.17 eV, below E F . Above E F these are observed from 0.10 eV up to 9.00 eV with an intragap between 0.91 eV and 3.30 eV. Down states appear from −8.55 eV up to −2.4 eV, followed for an intragap of 0.54 eV. At the Fermi level there is a continuous band from −1.80 eV up to 0.90 eV. Above E F , unoccupied states are observed from 1.00 eV up to 9.00 eV with an intragap between 2.50 eV and 3.20 eV.

Total DOS for (a) Sr2CoMoO6 and (b) Sr2CoReO6 compounds. In the former a metallic behavior is observed close to the Fermi level, while a half-metallic characteristic is determined for the latter
In the A2BB′O6 complex perovskite the effect of oxygen around the B and B′ transition metal cations produces a splitting of the crystalline field, which rises from \(d_{xy}, d_{xz}, d_{yz}, d_{z^{2}},d_{x^{2} - y^{2}}\) hybridization levels. It is known that for no cubic structures, d xz and d yz behave as a single level d xz+yz , which is a consequence of structure symmetry lost due the cubic cell deformation necessary to create a tetragonal structure.
Another explanation is related to the possibility that usual states are not divided in e g (high energy) and t 2g (low energy) but in \(d_{z^{2}}, d_{xy}\) (high energy) and \(d_{x^{2} - y^{2}}, d_{xz + yz}\) (low-energy) levels. Due to the magnetic characteristic of B and B′ cations, an exchange splitting has place. It is observed as a difference between e g up and down states, and a difference between t 2g up and down states. In order to study these effects, calculations of separate partial densities of Co, Mo have been performed for Sr2CoMoO6 and Co, Re for Sr2CoReO6. These were carried out by considering both up and down spin polarizations. Results are shown in Figs. 3 and 4.

Magnetic Partial DOS contribution of d orbital to Sr2CoMoO6 material: (a) distribution of Co-d and (b) Mo-d states for the up and down spin orientations. The z 2 and xy orbital contribution are superposed

Partial DOS contributions of d orbital to Sr2CoReO6 material: (a) distribution of Co-d and (b) Re-d states for the up and down spin orientations. The z 2 and xy orbital contributions are superposed
Figure 3a shows the DOS for the Co-3d levels of the Sr2CoMoO6. Note that \(d_{z^{2}}, d_{xy}\) orbital contributions are exactly superposed. Down polarized \(d_{x^{2} - y^{2}}, d_{xz + yz}\) levels give important contribution near the Fermi level. Meanwhile, \(d_{z^{2}}, d_{xy}\) levels belong to the unoccupied states. On the other hand, up orientated \(d_{z^{2}}, d_{xy}\) levels contribute to the conduction of material, while \(d_{x^{2} - y^{2}}, d_{xz + yz}\) are strongly localized below the Fermi level. Figure 3b exemplifies the DOS for the partial Mo-3d states of the Sr2CoMoO6. We determine that both up and down high-energy levels do not contribute to the total DOS close the Fermi level. This information permits to conclude that there are not states occupied by the Mo-4d level. The absence of Mo-4d levels implies that Mo has valence +6 and Co has +2 in the Sr2CoMoO6 compound and we do not expect a significant contribution of Mo to the cell magnetic moment.
Figure 4a exhibits partial densities for the Co-3d levels for the Sr2CoReO6 material. As in the previous case, \(d_{z^{2}},d_{xy}\) levels are equivalent and \(d_{x^{2} - y^{2}}, d_{xz + yz}\) states have a similar behavior. Down polarized low-energy states contribute to the conduction band, which appears from −1.80 eV up to 0.80 eV, while high-energy levels contribute to the unoccupied states above the Fermi level. On the other hand, up polarized high-energy states contribute below the Fermi level in the regime from −1.70 eV up to −0.20 eV and strongly localized below −3.15 eV (see Fig. 2). Low-energy states are observed below −3.00 eV. Figure 4b shows Re-5d levels.
It is observed that up and down high-energy states do not contribute significantly to the DOS and appear above 3.47 eV (see Fig. 2). Up low-energy states are located close and above the Fermi level at E>0.15 eV. We observed that down low-energy states principally belong to the conduction band. It is expected from the possible valences of Re, +5 or +6, which represent an effective magnetic moment due to 5d 1 or 5d 2 electronic configurations. The results of figure 4 show that conduction states are shared by the Co and Re cations, which suggests the hybridization of Re-5d and Co-3d levels.
Figure 5 shows the characteristic band structure of Sr2CoB′O6 (B′ = Mo, Re) materials. These results are absolutely in agreement with that of DOS. As observed in Figs. 5a and 5b, Sr2CoMoO6 evidences a metallic feature for both up and down spin orientations. In spite of the similarity between the band structure for Sr2CoMoO6 and Sr2CoReO6 compounds, the latter behaves as a half-metallic material, with a semiconducting behavior for the spin up channel (Fig. 5c) and conductor for the other one (Fig. 5d).

Band structure of Sr2CoB′O6 (B′=Mo, Re) for both up and down spin configurations. (a) Sr2CoMoO6 spin up, (b) Sr2CoMoO6 spin down, (c) Sr2CoReO6 spin up and (d) Sr2CoReO6 spin down
In order to explain the splitting of crystalline field and exchange, in Fig. 6 we present a qualitative scheme of DOS in Sr2CoMoO6 and Sr2CoReO6 materials for both up and down spin orientations. Figure 6a shows splitting for Sr2CoMoO6 material. In the case of Co, splitting of crystalline field (spin up) and exchange are more energetic. In the case of Mo the splitting of crystalline field is above 4.00 eV for both up and down spin configurations. For Mo no evidences of exchange splitting are observed because the high- and low-energy levels have the same electronic distributions. In the scheme of Fig. 6a, dark regions represent low-energy d states and clear regions high-energy d states; left for Co and right for Mo. It is clear that low-energy states contribute for the conduction band of material. There is no evidence of half-metallicity and the calculated effective magnetic moment due the exchange splitting between high- and low-energy states of Co is 2.71 μ B .

Qualitative scheme of splitting for Sr2CoB′O6 (B′=Mo, Re) compounds. (a) Distribution of Co-d and Mo-d states for the up and down spin configurations on Sr2CoMoO6 material, (b) distribution of Co-d and Re-d states for the up and down spin orientations, on the Sr2CoReO6 compound. Filled and dot lines represent minor and main energy states of each spin polarization, respectively
For Sr2CoReO6 material, in Fig. 6b we schematize the splitting of crystalline field with values of 0.92 eV for the spin up polarization of Co and 1.58 eV for spin down orientation. Splitting of exchange shows 0.42 eV for high-energy states and 1.88 eV for low-energy states. A breadth splitting of crystalline field, bigger than 5.00 eV of high-energy levels of Re for both up and down spin channels, reveals the unoccupied nature of these states. Low-energy states present a negligible splitting of exchange but it is clear that there is a magnetic moment which is related with these states around the Fermi level. The total magnetic moment was calculated to be 2.02 μ B due to the following contributions: p Sr=−0.004 μ B ; p Co=2.45 μ B ; p Re=−0.48 μ B ; p 0=0.03 μ B . As expected for half-metallic materials, the total cell magnetic moment is close to an integer number of Bohr magnetons. The presence of Re and Co states in the conduction band suggests the hybridization of these cations in order to make possible the origin of an electron hopping process. Then, we propose that Re spin down levels are displaced to the low-energy regime and Re spin up levels are displaced to the high-energy region, which supports the half-metallic behavior.
4 Conclusions
Ab initio studies of the electronic properties of Sr2CoB′O6 (B′=Mo, Re) complex perovskites were performed. The lattice parameters of the tetragonal perovskites were optimized by the minimization of energy as a function of the c/a ratio. The results are in agreement with experimental reports [6, 7]. The DOS introducing spin polarization shows that Sr2CoMoO6 presents a conductor behavior. On the other hand, the Sr2CoReO6 evidences a half-metallic nature, with a conductor characteristic for the spin down channel and semiconducting feature for the spin up orientation. The majority contribution to the conduction band for both metallic and half-metallic materials corresponds to the low-energy spin down states of Co. However, in the case of Sr2CoReO6, low-energy spin down states of Re give contributions to the conduction band. As expected for half-metallic systems [10], the effective magnetic moment of a cell was determined to be an integer number for the Sr2CoReO6 compound. It is predominantly due to the Co orbital.
References
Longo, J.M., Ward, R.: J. Am. Chem. Soc. 83, 1088 (1961)
Patterson, F.K., Moeller, C.W., Ward, R.: Inorg. Chem. 2, 196 (1963)
Kobayashi, K.I., Kimura, T., Saeada, H., Tekura, K., Tokura, Y.: Room-temperature magnetoresistance in an oxide material with an ordered double-perovskite structure. Nature 395, 677–680 (1998)
Garcia-Landa, B., Ritter, C., Ibarra, M.R., Blasco, J., Algarabel, P.A., Mahendiran, R., Garcia, J.: Magnetic and magnetotransport properties of the ordered perovskite Sr2FeMoO6. Solid State Commun. 110, 435–438 (1999)
Zhong, C., Fang, J., Jiang, Q.: Magnetodielectric effects in the ferroelectric ferromagnet BiMnO3. J. Phys., Condens. Matter 16, 9059–9168 (2004)
Kato, H., Okuda, T., Okimoto, Y., Tomioka, Y., Oikawa, K., Kamiyama, T., Tokura, Y.: Structural and electronic properties of the ordered double perovskites A 2 MReO6 (A=Sr,Ca; M=Mg,Sc,Cr,Mn,Fe,Co,Ni,Zn). Phys. Rev. B 69, 184412 (2004)
Viola, M.C., Martínez-Lope, M.J., Alonso, J.A., Velasco, P., Martínez, J.L., Pedregosa, J.C., Carbonio, R.E., Fernández-Díaz, M.T.: Induction of colossal magnetoresistance in the double perovskite Sr2CoMoO6. Chem. Mater. 14, 812–818 (2002)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kvasnicka, D., Luitz, J.: WIEN2k, An Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties. Karlheinz Schwarz, Techn. Universität Wien (2001), ISBN 3-9501031-1-2
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996)
Bonilla, C.M., Landínez Téllez, D.A., Arbey Rodríguez, J., Vera-López, E., Roa-Rojas, J.: Half-metallic behavior and electronic structure of Sr2CrMoO6 magnetic system. Physica B 398, 208–211 (2007)
Acknowledgements
This work was partially supported by Division of Investigations (National University of Colombia, Bogotá DC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bonilla, M., Landínez Téllez, D.A., Roa-Rojas, J. et al. Electronic Structure and Half-Metallic Character in Sr2CoB′O6 (B′ = Mo, Re) Materials. J Supercond Nov Magn 26, 2307–2312 (2013). https://doi.org/10.1007/s10948-012-1419-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-012-1419-2