
Abstract
The dissolution of intermediate lithium polysulfide within the electrolyte presents a significant challenge in lithium-sulfur batteries (Li–S). While an increasing number of recent studies on Li–S are focused on using activated carbon (AC) cathodes due to their strong affinity to lithium polysulfide, there still has been limited investigation into the quantitative adsorption of lithium polysulfide across various ratios of urea doping with AC. This study thus aims to quantitatively study the polysulfide adsorption capabilities of biomass-based carbon material activated by KOH and doped with urea. The correlation between N-doped surface morphology and lithium polysulfide in the AC was thoroughly investigated. The results indicate that increasing the urea ratio improves AC’s porosity and enhances the lithium polysulfide adsorption. AC prepared with the highest biomass: urea ratio of 1:3 in this study exhibits the most remarkable adsorption uptake capacity of 12.94 mmol/g. This excellent adsorption performance is attributed to the synergistic effect of well-developed porosity (BET surface area of 1902.99 cm2/g and a pore volume of 0.92 cm3/g) and high nitrogen functionalization on the carbon surface, contributing to the formation of physical and chemical bonds between polarized lithium polysulfide and the carbon matrix that enhances the adsorption process.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Lithium-sulfur (Li–S) batteries are promising candidates for the next-generation portable or stationary power sources, owing to their high theoretical energy density of up to 2600 Wh kg−1 [1, 2]. Considerable efforts have been undertaken to address the challenge of sulfur’s volumetric expansion (up to 80%) during lithiation to form lithium sulfide, as well as to enhance the inherently low conductivity of sulfur. Despite these efforts, the fast capacity degradation of Li–S batteries remains a significant problem. This is because highly soluble lithium polysulfide (LiP) intermediates (Li2Sx, 4 < x < 8) are generated and dissolved in liquid electrolytes during electrochemical reduction reactions (Fig. 1) [3]. These polysulfide ions then cyclically travel between the sulfur cathode and metallic lithium anode. In addition, the inefficient trapping of dissolved LiP by the weak physical adsorption of the cathode, coupled with the pronounced diffusion of LiP into electrolyte solvents [such as 1, 2-dimethoxyethane (DME) and 1,3-dioxolane (DOL)], promotes parasitic reactions and exacerbates the shuttle effect within Li–S batteries. Figure 1 provides a summary of LiP migration and its consequential shuttle effect. Further information regarding the migration of LiP and its resulting shuttle effect can be found elsewhere [4].

Migration of lithium polysulfide and shuttle effect
Various approaches have been employed to tackle the problems outlined above. These include incorporating functional interlayers between the cathode and separator [5], modifying the separator [6], and developing a novel electrolyte [7]. Among these endeavors, using a nitrogen-functionalized activated carbon (AC) as a cathode provides the most facile ways to anchor LiP. AC has high electronic conductivity, which allows it to effectively mitigate the electrical insulation of sulfur and its discharge products [8]. Furthermore, the substantial pore volume of AC can accommodate the volume change of sulfur cathodes throughout the cycling process [9, 10]. In addition, through the incorporation of nitrogen, the poor affinity between non-polar carbon and polar LiP can be improved. Nitrogen, which has a relatively high electronegativity (EN = 3.04), provides free electrons, resulting in increased charge densities on adjacent carbon atoms, thereby polarizing the surface of the carbon atom [11, 12]. Furthermore, recent studies have indicated that hierarchical porous carbon with a substantial specific surface area can effectively confine the migration and undesirable shuttle reactions of LiP within micropore structures. Simultaneously, it facilitates the movement of lithium ions through the mesopores and provides adequate surface area for electrochemical reactions [13].
Despite representing a significant advancement, there remains a lack of comprehension regarding the influence of the nitrogen doping level and hierarchical structures on the inhibition of LiP. To the best of our knowledge, only a limited number of studies have quantitatively evaluated the LiP adsorption capacity on the surface of nitrogen-doped AC (as opposed to the typical electrochemical analysis). Therefore, further investigations are imperative to explore LiP uptake and comprehend the mechanisms underlying LiP adsorption. Driven by these considerations, this study aims to quantitatively study the LiP adsorption capabilities of different ACs from palm kernel shells (PKS) prepared by potassium hydroxide (KOH) activation and varying nitrogen doping ratios. PKS possesses a robust mechanical structure and a considerably high nitrogen content, which are good features of AC. The findings revealed that the deliberately designed AC structure exhibits a highly polarized surface chemistry, with a substantial surface area and an abundance of hierarchical pores. These distinctive properties prove effective in suppressing LiP and show great potential for the N-doped PKS-derived AC as a promising cathode material in Li–S battery applications.
2 Materials and methods
2.1 Preparation of nitrogen-doped activated carbon
Nitrogen-doped activated carbon (PSC-Ux, x = 1,2,3) was prepared by carbonization-activation step using PKS as a starting biomass material and urea as the nitrogen source. PKS was acquired from the Sime Darby Research and Development Center (R&D) in Carey Island, Malaysia. The PKS was first rinsed with distilled water and dried overnight in an oven at 110 °C. The dried PKS was then crushed and sieved to obtain particle sizes of 0.15 to 0.2 mm. The PKS was mixed with 1 M KOH (Analytical grade, ChemAr) and urea (1:1:1), agitated for three hours with a magnetic stirrer, and desiccated at 110 °C to remove water. Subsequently, the mixture was placed in an electric tube furnace (OTF-1200, MTI Corporation, USA) and annealed under N2 (99% pure, Linde Malaysia Sdn. Bhd.) flows at 800 °C for 2 h at a rate of 10 °C/min. The activated carbon (AC) obtained was washed under stirring with 1 M HCl solution (85 wt%), followed by hot distilled water until the pH reached 7, and dried at 110 °C for 24 h. The resultant AC was designated as PSC-U1. PSC-U2 and PSC-U3 were produced using the same synthesis method as PSC-U1, but with different mass ratios of PKS to urea, specifically 1:2 and 1:3, respectively. For comparison purposes, a version of AC (PSC) was prepared without the inclusion of urea.
2.2 Characterization and surface functionalities analysis of activated carbon
The specific area and pore volume were measured according to the Brunauer–Emmett–Teller (BET) method at 77 K, using a Quantachrome Autosorb Automated Gas Adsorption system. The external structure and elemental material were observed using a scanning electron microscopy (SEM-EDX); (Zeiss/Auriga FIB). The functional groups of all samples were analyzed using a Fourier transform infrared (FTIR) spectroscopy (Perkin Elmer Spectrum One FTIR spectrometer). The crystalline materials were characterized by X-ray diffraction (XRD) using a Bruker D8 Advance (Bruker Corporation, Germany). X-ray photoelectron spectroscopy (XPS); (Kratos/Shimadzu) was used to analyze the elemental composition. The Raman spectrum was measured using a Raman analyzer (LabRAM HR Evolution) with a 532 nm blue laser. Elemental analyses were performed using a CHNS Elemental Analyzer 2400 Series II PerkinElmer instrument.
2.3 Preparation of lithium polysulfide
High-order lithium polysulfides, Li2S6 was prepared by chemically reacting sublimed sulfur and Li2S in a 5:1 molar ratio in a DOL/DME solution (1:1 by volume). The solution was constantly stirred at 600 rpm and 70 °C in an Argon-filled glove box overnight to produce a brownish–red Li2S6 solution (0.2 M). The LiP was then diluted several times to obtain a calibration curve. A concentration of 15.0 mM was used for the polysulfide adsorption test. A blank containing no LiP was used as a control, and a calibration curve of absorbance versus concentration was constructed using a UV–Vis spectrophotometer (Cary 60, Agilent Technologies).
2.4 Adsorption of lithium polysulfide, Li2S6
10 mg of each AC was placed in 10 mL of LiP solution and allowed to rest for 60 min in an argon-filled glove box. Subsequently, the samples were centrifuged at 200 rpm for 10 min before undergoing UV–Vis spectrophotometer analysis to measure their adsorption capacity. To ensure the reliability of the data, three sets of repetitions were conducted. The adsorption capacity was determined by Eq. (1):
where, qt is LiP uptake, (mmol/g), Co is the LiP initial concentration (mg/L), Ct is the LiP concentration at time t (mg/L), m is the mass of AC (g) and V is the volume of solution.
It should be noted that extreme caution is necessary throughout LiP adsorption. This is because LiP is susceptible to disproportionation and may reduce into Li2S4 or Li2S8, which can potentially impact the outcome of quantitative and qualitative results [14]. Furthermore, LiP is unstable in the ambient environment; and can lead to color fading, thereby nullifying the validity of the analysis. Thus, the samples must be prepared and conducted with proper sealing in an argon environment and using amber glass to mitigate the disproportion of Li2S6.
3 Results and discussion
3.1 Proximate and ultimate analyses
Table 1 presents the results of the proximate analysis, revealing that raw PKS has a proportion of volatile matter, fixed carbon, ash, and moisture of 68.94, 18.75, 4.92 and 7.39 per cent (wet basis), respectively. Subsequent to undergoing pyrolysis with KOH treatment, all samples, irrespective of the urea doping level, showed a decrease in volatile content due to the degradation of hemicellulose, cellulose, and lignin components, which continuously release oxygen, hydrogen, and carbon during structural rearrangement [15, 16]. Notably, the amount of fixed carbon, corresponding to the pure carbon of all ACs, increases significantly. In addition, the ash’s inorganic constituents (mineral elements) appeared to be stable and less susceptible to decomposition under high activation temperature, resulting in a minor increase in the resultant PSC-Ux (x = 1,2,3).
The results of the ultimate analysis of raw PKS, PSC, and PSC-Ux are also presented in Table 1. The data indicate the successful introduction of nitrogen onto the surface of AC through the activation reaction with urea. Among the samples, PSC-U3 exhibited the highest nitrogen content, while PSC had the lowest due to the poor stability of the inherent nitrogen-containing functional groups, which were released at high temperatures [17]. The hydrogen and oxygen contents of PKS decreased due to the dehydration and decarboxylation by KOH and were eliminated as volatile compounds in the form of liquid condensate or vapor [18]. Additionally, the hydrogen content could have been further diminished by aromatization and the cleavage of heavy-molecular-weight hydrocarbons at high temperatures [19]. Notably, the sulfur content in both raw PKS and ACs was deemed negligible and fell below the detection limit.
3.2 Examination of AC surface modification from various analysis
The capacity of AC to confine LiP within its pore structure correlates well with the surface development, surface area, pore volume, and types of pores. Moreover, given that LiP in DME/DOL is regarded as a polar species, the AC surface is nitrogen-doped with nitrogen to induce surface polarization, consequently fostering more robust chemical interactions with LiP. This section discusses the modification of the surface morphology and surface chemistry of the PSC and PSC-Ux through SEM-EDX, XRD, Raman, N2 adsorption, BET surface area, and XPS analyses.
3.2.1 SEM-EDX analysis
The SEM-EDX analysis results for the PSC and PSC-Ux are shown in Fig. 2. All the samples exhibited a porous network structure with various cavities. The formation of rough surfaces can be attributed to the degradation of lignocellulosic precursors at elevated temperatures, causing the evaporation of volatile compounds and the generation of newly formed pores [20]. Furthermore, KOH impregnation facilitated the formation of pores through the intercalation of metallic potassium into the carbon lattice, which extended the carbon layers [4, 21]. The effect of variations in the urea doping ratio on the morphology shown in Fig. 2b–d indicates that the pore structure of the modified activated carbon was formed with interconnected pore clusters. The addition of heteroatom doping led to a more varied texture with an irregular surface morphology. Meanwhile, the morphology of the PSC (Fig. 2a) showed an abundance of microporous structures. The presence of cavities is essential for providing van der Waals forces to LiP through physical adsorption. The EDX analysis was used to quantify the elemental composition of the activated carbon. The results presented in Fig. 2 show that an increase in urea has a favorable impact on the nitrogen content in the AC. These results are consistent with the proximate analysis.

SEM imagery and EDX analysis of PSC, PSC-U1, PSC-U2 and PSC U3
3.2.2 X-ray diffraction and Raman spectroscopy
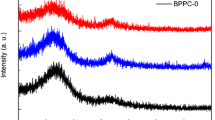
Figure 3a demonstrates the presence of broad peaks at 25° and 42°, indicating the decomposition of the organic material of amorphous ACs. The peak intensity is denoted by the atomic position of the crystal structure. PSC exhibits significant intensity due to the heightened periodicity in any other direction or preferable crystal orientation [22]. Notably, the intensity of the diffraction peaks decreases with PSC-Ux, suggesting that the inclusion of urea leads to the collapse of layered graphitic planes and further exacerbates the degree of amorphousness [23, 24]. Raman shift of PSC and PSX-Ux are shown in Fig. 3b. The two prominent peaks centered at 1340 cm−1 and 1580 cm−1 are attributed to the D-band and G-bands, respectively. The G-band corresponds to the first-order stretching vibration of unsaturated carbon, whereas the D-band is related to the disruption of symmetry in sp2-hybridized carbons, leading to structural defects [25]. The D-band to G-band (ID/IG) ratio represents the carbon material’s graphitization degree [26]. In general, the addition of monoatomic or heteroatomic doping into the synthesis of PSC-Ux increases the disorder of carbon structures. This is evidenced by the highest ID/IG ratio (1.0) for PSC-U3, indicating the least degree of crystallization.

a X-ray diffraction pattern and b Raman spectra of PSC, PSC-U1, PSC-U2 and PSC-U3
3.2.3 N2 adsorption
Porous structures with a high surface area and pore volume have the potential to adsorb a substantial amount of LiP. The variations in porosity properties between PSC and PSC-Ux were assessed through N2 adsorption/desorption isotherm analysis. As illustrated in Fig. 4a, all carbon materials displayed type I/IV isotherms, according to the IUPAC classification, with hysteresis loops at the middle of relative pressure (P/Po), implying that micropores and mesopores (hierarchical) structures co-exist in these samples [27, 28]. The synergetic effect of chemical activation of KOH and high activation temperature primarily causes the hierarchical porous structure. This is due to the production of CO2 from the decomposition of K2CO3 (Eq. 2) at 800 °C, which contributes to further porosity development via carbon gasification [29, 30]. Simultaneously, K2CO3 and K2O are also reduced into metallic K (Eqs. 3, 4) that can intercalate into the carbon lattices and expand irreversibly upon activation [31]. This led to the creation of micropores and mesopores structures for the LiP entrapment.

a Nitrogen adsorption–desorption isotherms and b pore size distribution of PSC, PSC-U1, PSC-U2 and PSC-U3
Data in Table 2 indicate that the BET surface area and pore volume of PSC-Ux were higher than the PSC. A comparison of porosity development across varying levels of urea doping suggests that PSC-U3 has the highest BET surface area of 1902.99 cm2/g and pore volume of 0.92 cm3/g. Urea plays a significant role in enhancing the specific surface area and porosity of N-doped AC by exerting an additional impact on altering the porous structure. Upon incorporation into the AC matrix, urea introduces defects and nitrogen functional groups onto the surface. These defects create active sites that are capable of interacting with LiP molecules. During the activation process, urea infiltrates into the PKS fiber and interacts with other functional groups, such as –OH and –COOH from cellulose and lignin, forming –N–H or –N–C bonds, causing significant expansion of the fiber [32,33,34]. These interactions facilitate pore opening and create active sites for LiP adsorption. Moreover, nitrogen atoms (0.092 nm) are slightly larger than carbon atoms (0.0914 nm), and their inclusion can result in an expanded interlayer spacing, creating more space, and further increasing the available surface area [35, 36]. XRD and Raman analyses provide further evidence that high concentration of urea disrupts the surface of AC, leading to the development of a graphite-like surface that facilitates the formation of a porous structure. The pore size distribution (Fig. 4b) further proves the hierarchical porosity of PSC and PSC-Ux, with a wide pore distribution from mesopores to micropores ranging from 1 to 3 nm. The proportion of mesoporous structures becomes more prevalent with increasing urea doping. A similar explanation in terms of modification on the carbon surface by heteroatom doping applies to this observation.
3.2.4 XPS analysis
X-ray photoelectron spectroscopy (XPS) was employed to examine the chemical composition, bonding characteristics, and elemental states of the samples. The wide-survey XPS spectrum (Fig. 5a) displays three distinctive peaks centered at 284, 399, and 531 eV, corresponding respectively to C 1s, N 1s, and O 1s [37]. PSC-Ux consists mainly of carbon and oxygen, with a minor presence of nitrogen atoms. In contrast, PSC is solely composed of carbon and oxygen atoms. This underscores the successful incorporation of nitrogen into the surfaces of PSC-Ux materials. The nitrogen content in PSC-U1 (0.58%), PSC-U2 (0.86%), and PSC-U3 (1.07%) increases proportionally with the urea content. The thermal decomposition of urea and KOH results in the generation of NH3 (Eq. 5). Subsequently, NH3 interacts with carbon atoms at the edges and peripheries of aromatic rings or defect sites associated with unpaired electrons. The carbon atoms possess high potential energy and react with NH3 into heterocyclic nitrogen structures to produce pyrrolic, pyridinic, and graphitic nitrogen on the surface of AC [38, 39]. The presence of pyridinic-N (398.4 eV), pyrrolic-N (399.5 eV), and graphitic-N (401.1 eV) is confirmed by high-resolution N 1s spectra, as shown in Fig. 5b–d. Pyridinic-N and pyrrolic-N serve as electron-rich donors capable of forming robust chemical interactions with polar LiP (Li+) [40]. As indicated in Table 3, pyridinic-N and pyrrolic-N predominate in PSC-U2 and PSC-U3, which are advantageous for capturing LiP.

a Wide XPS spectra for all samples and high-resolution N 1s spectrum of b PSC-U1, c PSC-U2, and d PSC-U3
3.3 Polysulfide adsorption
For the quantification of LiP adsorption capacity, a linear calibration curve was established, encompassing concentrations ranging from 0.8 mmol/L to 15 mmol/L (Figure S1). This concentration range is in line with the recommendations by Happauf et al. [41] for a battery system. Subsequently, the LiP adsorption assessment was conducted on PSC and PSC-Ux samples. In this procedure, 0.1 g of each sample was introduced into 10 ml of LiP solution, and the mixture was allowed to settle for 60 min within the glove box. Figure 6a shows that the PSC-U3 recorded the highest LiP adsorption capacity of 12.94 mmol/g, surpassing PSC-U2, PSC-U1, and PSC, with respective capacities of 9.48, 4.51, and 0.73 mmol/g. The superior LiP adsorption in PSC-U3 can be attributed to its well-developed porosity and the nearly equivalent distribution of micro- and mesoporous structures when compared to the other samples. This distinction is clearly evident in Table 2, where the BET surface area and total pore volume are approximately 30% greater than those of other ACs. Moreover, the substantial nitrogen content within PSC-U3 significantly contributes to enhanced LiP adsorption. This is facilitated by the immobilization of LiP through strong coordination interactions and chemical bonding with nitrogen moieties [42].

a Li2S6 polysulfide adsorption test, b continuous UV–Vis spectrum of Li2S6 adsorption and c visual comparison of final concentration of all samples
The polarity of the adsorbent plays a pivotal role in influencing the LiP adsorption. As discussed earlier, LiP in solution possesses a distinct polar adsorptive nature. Hence, relying solely on a physical adsorption mechanism proves to be relatively inefficient. Notably, even though PSC-U1 and PSC-U2 exhibit comparable surface areas and pore volumes (Table 2), the adsorption behavior of LiP on these surfaces differs greatly. Remarkably, the adsorption of LiP on PSC-U2 was approximately twice that observed on PSC-U1. This substantial enhancement in LiP adsorption capacity can be attributed to the greater degree of nitrogen functionalization in PSC-U2. The presence and composition of pyridinic-N and pyrrolic-N groups significantly contribute to a robust chemical adsorption capability within PSC-U2. These results strongly validate that, during operation, the confinement of LiP within the framework is primarily facilitated by robust chemical bonds rather than mere physical attraction.
Continuous UV–Vis absorbance was recorded within the range of 200 to 500 nm for all ACs after adsorption. In general, higher LiP concentrations absorb higher wavelengths and have a stronger absorbance uptake [43]. The performance of PSC and PSC-Ux in the LiP adsorption test is depicted in Fig. 6b. PSC demonstrates a notably high UV absorbance, indicative of a higher final concentration of adsorbed LiP. This suggests that PSC has a relatively low adsorption capacity. Conversely, as the urea doping increases, the UV–Vis absorption decreases, implying that the nitrogen functional group plays a crucial role in the binding of LiP during the adsorption process. Notably, PSC-U3 exhibits the lowest UV–Vis absorbance capacity, further affirming its strong capability in adsorbing LiP.
3.4 Proposed LiP adsorption mechanism
The mechanism underlying the adsorption of LiP onto the surface of ACs is primarily influenced by surface morphology and surface chemistry, involving various interactions such as physisorption and chemisorption. Surface morphology refers to the physical structure modification of AC that generates suitable proportions of micro, meso, or hierarchical porous structures. Surface chemistry, on the other hand, involves the incorporation of doping elements onto the surface, thereby instigating diverse chemical interactions such as nitrogen bonding, Lewis’s acid-based interaction, and π–π conjugation. These interactions synergistically enhance the chemical reaction between LiP and the ACs [44, 45].
PSC-U3 showcases a highly advantageous porous structure imbued with noticeable structural imperfections, leading to a substantial increase in surface area and a larger pore volume. These defective surfaces offer ample active sites for the entrapment of LiP molecules within the carbon matrix. Table 2 provides clear evidence of a significant decrease in specific surface area and pore volume of PSC-U3-AA by 67% and 70%, respectively, demonstrating the remarkable adsorption capacity of LiP after 60 min. The mechanism of LiP adsorption onto PSC-U3 involves several stages. Initially, LiP molecules diffuse from the bulk solution to a film layer of PSC-U3 particles via molecular diffusion, followed by diffusion from the film layer to the particle surface (film diffusion). Eventually, the LiP molecules migrate inside the PSC-U3 through pore diffusion [46]. Additionally, PSC-U3 exhibits hierarchical porous structures, finely balanced between micropores (55.43%) and mesopores (44.57%), providing an ideal spatial arrangement for LiP adsorption. This is of significance, as the pore width must be larger than the size of LiP molecules, approximately 0.8 nm, for efficient adsorption to take place [47, 48]. The appropriate distribution of pore sizes plays an important role in creating a strong Van der Waals force between LiP molecules and the adsorbent material. When LiP molecules pass through a suitable pore size, their energy gradually decreases. Upon entrainment, the interactions between the LiP molecules and the pore walls are governed by the Lennard-Jones potential [49]. Eventually, the LiP molecules get tightly packed within the carbon’s framework.
As indicated by the XPS analysis, the presence of highly doped nitrogen (pyridinic-N and pyrrolic-N with lone pair electrons) in the PSC-U3 facilitates chemisorption by the interaction of the Lewis acid-based terminal Li atoms in LiP [43]. These nitrogen-functionalized surfaces form a π-π conjugated structure, which can accept additional π electrons, thus further enhancing the dipole-dipole interaction to form a strong Li–N bond through chemical interactions and imparting a polar surface, enabling them to chemically confine LiP on the carbon surface [50] This is supported by the prominent intensity peaks of FTIR (Fig. 7) observed in PSC-U3-AA at 3412 cm−1, 1572 cm−1, 1097 cm−1, and 1012 cm−1, which indicate the involvement of the functional groups of PSC-U3 in LiP adsorption. The peaks at 3412 cm−1 suggest an interaction between LiP and the N–H stretching of the amine group. Additionally, the peak at 1572 cm−1 is attributed to a chemical bond between Li+ and the nitrogen functional groups (primarily pyrrolic-N) [51], while the peaks at 1097 to 1012 cm−1 indicate the π-π attraction due to the presence of π-electrons in both the adsorbate (LiP) and the adsorbent (PSC-U3) [52]. However, in the case of graphitic-N, there are no additional lone pair electrons available to anchor LiP. Instead, graphitic-N functions as a mono-functional site by utilizing all of its π electrons to form a π-conjugated system [53]. This arrangement helps enhance the charge density within the doping regions. The synergistic impact of these interactions significantly contributes to the substantial adsorption capacity. A schematic illustration of the proposed mechanism and adsorption process is provided in Fig. 8.

FTIR spectra of PSC-U3 (before adsorption) and PSC-U3-AA (after adsorption)

Proposed mechanism of physical and chemical adsorptions of LiP on AC
4 Conclusions
A comprehensive investigation was undertaken to quantitatively assess the adsorption of lithium polysulfides using activated carbon with varying degree of urea doping. Nitrogen-doped activated carbon (PSC-Ux) was successfully synthesized by subjecting palm kernel shell to KOH activation at elevated temperatures. The porosity analysis revealed that PSC-U3 exhibited the highest surface area of 1902.99 m2/g and a large total pore volume of 0.92 cm3/g, with a nearly equal distribution of microporous and mesoporous structures. The incorporation of nitrogen doping (pyridinic-N, and pyrrolic-N) on the surface of activated carbon enhanced the chemical binding capacity. This can be observed in PSC-U3, which had the highest nitrogen content of 3.26 wt%, demonstrating superior lithium polysulfide adsorption of 12.94 mmol/g. The adsorption mechanism involved physical adsorption through van der Waals forces and chemical adsorption via Lewis’s acid-based and π–π interactions. These findings offer invaluable insights for the development of excellent activated carbon cathodes for Li–S battery application.
References
J.S. Yeon, S.H. Park, J. Suk, H. Lee, H.S. Park, Chem. Eng. J. 382, 1 (2020)
Y.-W. Song, J.-L. Qin, C.-X. Zhao, M. Zhao, L.-P. Hou, Y.-Q. Peng, H.-J. Peng, B.-Q. Li, J. Energy Chem. 64, 568 (2022)
J.E. Knoop, S. Ahn, J. Energy Chem. 47, 86 (2020)
M.S.M. Zaini, N. Faradila, S. Abdul, M. Al-junid, S.S.A. Syed-hassan, Mater. Sci. Energy Technol. 6, 205 (2023)
Q. Hu, J. Lu, C. Yang, C. Zhang, J. Hu, S. Chang, H. Dong, C. Wu, Y. Hong, L. Zhang, Small. 16, 2002046 (2020)
J. Xu, S. An, X. Song, Y. Cao, N. Wang, X. Qiu, Y. Zhang, J. Chen, X. Duan, J. Huang, Adv. Mater. 33, 2105178 (2021)
B. Liu, Y. Zhang, Z. Wang, C. Ai, S. Liu, P. Liu, Y. Zhong, S. Lin, S. Deng, Q. Liu, Adv. Mater. 32, 2003657 (2020)
F. Chen, J. Yang, T. Bai, B. Long, X. Zhou, Electrochim. Acta. 192, 99 (2016)
F. Li, R. Liu, V. Dubovyk, Q. Ran, B. Li, Y. Chang, H. Wang, H. Zhao, S. Komarneni, Food Chem. 366, 130563 (2022)
H. Zhao, G. Zhu, F. Li, Y. Liu, M. Guo, L. Zhou, R. Liu, S. Komarneni, Sens. Actuators B Chem. 380, 133309 (2023)
Z. Song, X. Lu, Q. Hu, J. Ren, W. Zhang, Q. Zheng, D. Lin, J. Power Sources. 421, 23 (2019)
L. Kong, B. Li, H. Peng, R. Zhang, J. Xie, J. Huang, Q. Zhang, Adv. Energy Mater. 8, 1800849 (2018)
J. Park, S. Yoon, S. Oh, J. Kim, D. Kim, G. Kim, J. Lee, M.J. Song, I. Kim, K. Sohn, J. Kim, J. Energy Chem. 61, 561 (2021)
D.S. Wu, F. Shi, G. Zhou, C. Zu, C. Liu, K. Liu, Y. Liu, J. Wang, Y. Peng, Y. Cui, Energy Storage Mater. 13, 241 (2018)
M.S.M. Zaini, S.S.A. Syed, Hassan, Malaysian J. Anal. Sci. 20, 1390 (2016)
M.M. Nurfarhana, N. Asikin-Mijan, S.F.M. Yusoff, Mater. Chem. Phys. 308, 128196 (2023)
R. Pietrzak, H. Wachowska, P. Nowicki, Energy and Fuels. 20, 1275 (2006)
M. Ameen, N.M. Zamri, S.T. May, M.T. Azizan, A. Aqsha, N. Sabzoi, F. Sher, Biomass Convers. Biorefinery. 12, 103 (2022)
E. Yi, X. Shen, X. Chen, M. Jiang, W. Yan, Y. Liu, W. Jiang, M. Abdullah, Chem. Eng. Sci. 282, 119193 (2023)
H. Rustamaji, T. Prakoso, H. Devianto, P. Widiatmoko, W.H. Saputera, J. Energy Storage 52, 104724 (2022)
C.-C. Wang, Y.-W. Lin, S.-M. Lee, Surf Coat. Technol. 400, 126228 (2020)
M. Guo, F. Li, Q. Ran, G. Zhu, Y. Liu, J. Han, G. Wang, H. Zhao, Microchem J. 190, 108709 (2023)
X. Wu, C.W.K. Lam, N. Wu, S.-S. Pang, Z. Xing, W. Zhang, Z. Ju, Mater. Today Energy. 11, 182 (2019)
H. Zhao, Y. Liu, F. Li, G. Zhu, M. Guo, J. Han, M. Zhao, Z. Wang, F. Nie, Q. Ran, Ceram. Int. 19, 37549 (2023)
X. Lang, X. Wang, Y.Y. Li, K. Cai, L. Li, Q. Zhang, Sustain. Energy Technol. Assess. 43, 100947 (2021)
M. Xiang, Y. Wang, J. Wu, Y. Guo, H. Wu, Y. Zhang, H. Liu, Electrochim. Acta. 227, 7 (2017)
Y. Sun, D. Xu, S. Wang, Carbon N. Y. 199, 258 (2022)
M.S.M. Zaini, M.J. Jalil, Kem. u Ind. 70, 717 (2021)
M. Xue, C. Chen, Y. Tan, Z. Ren, B. Li, C. Zhang, J. Mater. Sci. 53, 11062 (2018)
M.S.M. Zaini, M. Arshad, S.S.A. Syed-Hassan, J. Bioresour Bioprod. 8, 66 (2022)
G. Singh, J. Lee, R. Bahadur, A. Karakoti, J. Yi, A. Vinu, Chem. Eng. J. 433, 134464 (2022)
J. Ren, Y. Zhou, M. Guo, Q. Zheng, D. Lin, Int. J. Hydrogen Energy. 43, 20022 (2018)
R. Ma, J. Hao, G. Chang, Y. Wang, Q. Guo, Can. J. Chem. Eng. 98, 503 (2020)
Z. Cai, B. Liu, X. Zou, H.M. Cheng, Chem. Rev. 118, 6091 (2018)
S. Mukherjee, T.P. Kaloni, J. Nanoparticle Res. 14, 1 (2012)
Z. Zhang, X. Du, J. Wang, W. Wang, Y. Wang, Z. Fu, Powder Technol. 254, 131 (2014)
H. Feng, M. Zhang, J. Kang, Q. Su, G. Du, B. Xu, Mater. Res. Bull. 113, 70 (2019)
S. Wang, K. Zou, Y. Qian, Y. Deng, L. Zhang, G. Chen, Carbon N. Y. 144, 745 (2019)
N. Tsubouchi, M. Nishio, Y. Mochizuki, Appl. Surf. Sci. 371, 301 (2016)
K. Yang, J. Yan, R. He, D. Li, Y. Li, T. Li, B. Ren, J. Electroanal. Chem. 861, 113922 (2020)
F. Hippauf, W. Nickel, G.P. Hao, K. Schwedtmann, L. Giebeler, S. Oswald, L. Borchardt, S. Doerfler, J.J. Weigand, S. Kaskel, Adv. Mater. Interfaces. 3, 1 (2016)
M. Zhang, J. Mu, Y. Li, Y. Pan, Z. Dong, B. Chen, S. Guo, W. Yuan, H. Fang, H. Hu, J. Energy Chem. 78, 105 (2023)
M. Zhong, J. Guan, J. Sun, H. Guo, Z. Xiao, N. Zhou, Q. Gui, D. Gong, Electrochim. Acta. 299, 600 (2019)
Z. Barlow, Z. Wei, R. Wang, Mater. Chem. Phys. 309, 128316 (2023)
M. Cao, Y. Feng, P. Zhang, L. Yang, X. Gu, J. Yao, J. Alloys Compd. 907, 164499 (2022)
Y. Li, H. Chen, K. Lim, H.D. Deng, J. Lim, D. Fraggedakis, P.M. Attia, S.C. Lee, N. Jin, J. Moškon, Nat. Mater. 17, 915 (2018)
C. Hu, H. Chen, Y. Shen, D. Lu, Y. Zhao, A.H. Lu, X. Wu, W. Lu, L. Chen, Nat. Commun. 8, 1 (2017)
M.S.M. Zaini, N.H.A. Rani, N.F. Mohamad, N.H. Sabri, Proc. 1St Int. Conf. Chem. Sci. Eng. Technol. 2703, 050003 (2023)
E. Troschke, C. Kensy, F. Haase, S. Dörfler, Y. Joseph, B.V. Lotsch, S. Kaskel, Batter. Supercaps. 3, 1069 (2020)
D. Wu, J. Chen, W. Zhang, W. Liu, J. Li, K. Cao, Z. Gao, F. Xu, K. Jiang, J. Colloid Interface Sci. 579, 667 (2020)
Y.F. Jia, B. Xiao, K.M. Thomas, Langmuir. 18, 470 (2002)
M. Saxena, N. Sharma, R. Saxena, Surf. Interfaces 21, 100639 (2020)
H. Zhang, Z. Zhang, J. Luo, X. Qi, J. Yu, J. Cai, Z. Yang, ChemSusChem 12, 283 (2019)
Acknowledgements
This study was supported by the Malaysian Ministry of Higher Education (MOHE) through the Fundamental Research Grant Scheme (FRGS/1/2022/TK0/UITM/02/82). M.S.M. Zaini is grateful to Universiti Teknologi MARA and the Ministry of Higher Education Malaysia for the scholarship award.
Author information
Authors and Affiliations
Contributions
MSMZ: Conducted the investigation, carried out formal analysis, contributed to the development of the methodology and conceptualization, and authored the initial draft of this manuscript. SAMAJ: Provided overall supervision for the experiment and performed validation on the results. SSAS-H: Supervised the experiment, contributed to manuscript writing, reviewing, and editing, and played a crucial role in acquiring funding.
Corresponding author
Ethics declarations
Conflict of interest
The authors assert that they have no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Md Zaini, M.S., Al-Junid, S.A.M. & Syed-Hassan, S.S.A. Effects of nitrogen doping on lithium polysulfide anchoring by activated carbon derived from palm kernel shell. J Porous Mater 31, 831–842 (2024). https://doi.org/10.1007/s10934-024-01556-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-024-01556-1