
Abstract
In this paper, the synthesis of core shell structured NiO@MCM-41 nanocomposite via vesicles as soft template is reported for the first time. Its catalytic performance was investigated in the CO2 reforming of methane (CRM) conversion. Stable vesicles first formed with CTAB/SDBS surfactant ratio of 1:2. Nickle nitrate was added to the vesicle mixture followed by addition of the aqueous solution of vesicle containing Ni cations inside to the MCM-41 gel. After high-temperature calcination, NiO@MCM-41 nanocomposite were obtained. The structural symmetry and the surface morphology were characterized by transmission electron microscope (TEM), low angle X-Ray Diffraction (XRD) and N2 adsorption/desorption analysis. TEM image confirmed core–shell structure and the hexagonally ordered structure of shell of MCM-41 silica. The results indicated that the average diameter of synthesized core–shell NiO@MCM-41 particles is 70–80 nm and the most of them are of spherical shape. The result of small angle XRD and N2 isotherm adsorption/desorption analyses indicated successfull formation of mesoporous shell. Hydrogen consumption by the catalyst mainly at 700 °C in TPR profile showed the strong interaction of the most of Nickel content with the support. CRM conversion on the prepared catalyst after 245 min of reaction led to H2 conversion at 42%, CO2 conversion at 48% with H2/CO yield ratio of 0.8.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Nowadays significant efforts to design and synthesis of nanomaterials with controlled properties are applied, resulting in the development of nanocomposites such as bimetallic, the core–shell or multi-layered. Similar to other nanomaterials, many processes in the preparation of these composites take place [1]. Particle properties are often controlled by coating or encapsulating them in a selective shell [2,3,4,5,6]. For some applications, control of the morphology and size of the coating shell has been done by using a template. The shell can influence load, performance and reactivity level and can increase stability and dispersion of colloidal core. For example, covering the colloids in a shell of different combinations can protect them from physical and chemical changes in the drug delivery [7,8,9]. Recently, core–shell nanostructures consisting of metals and metal oxides have become an attractive research area in material chemistry. Core–shell composites often show the practical physical and chemical properties superior to their single counterparts, and thus potentially are useful in a wider range of applications such as electronics [10, 11] and medicine for controlled drug delivery [12,13,14], Bio-medical imaging [15, 16], As well as in tissue engineering [17, 18], biosensors [19, 20], cell labelling [21], and catalysis [20, 24, 25, 27, 28]. This combination of core and cladding material show different properties from the composition of the core and the shell alone. The optimization of the specific characteristics of particles through the coating process is the goal of many researchers. The core ingredients and layer can be a polymer, inorganic compounds and metals. The core–shell nanocomposite have shown high potential to improve the core with the suitable shell to achieve the specific catalytic performance [22,23,24,25,26]. In this way not only the activity of the catalyst is largely preserved, but also the consumption of precious metals is also greatly saved. Core–shell structure has better resistance compared to the metal when directly deposits on the support surface, because shell as a microcapsule, acts like a selector and upgrades the adsorption of reactants to the core metal particles and on the other hand prevents sintering of metal particles at high temperatures [27]. The synthesis of ordered SiO2 shell remains a challenge, because of incomplete cover of cores, multiple oxide domains in each zone, or particle aggregation to large clusters. The methods using vesicles as template provide a powerful strategy to control the size and morphology of core–shell composites [28, 29]. Vesicles of anionic and cationic surfactants in aqueous solution have received a great interest for a wide range of applications including colloids, pharmaceuticals, catalysts and materials with their use as simple model systems for biological membranes [30,31,32]. One of the applications of the core–shell structure is in the research studies of the catalytic dry reforming of methane [10, 33,34,35,36]. Reports indicate that the transition metals with the exception of osmium (Os); nickel (Ni), ruthenium (Ru), rhodium (Rh), Palldium (Pd), iridium (Ir) and platinum (Pt) can be used as catalysts in the dry reforming of methane [37, 38]. Among these metals Rh and Ru are the most active and strongest catalyst against coking [39, 40]. However, due to limited availability and high cost, these metals are not of economic justification [38]. On the other hand, nickel catalysts were noted due to the low cost and availability [41]. But they also have some problems such as agglomeration of nickel particles and the possibility of carbon deposition on the catalyst surface or in a reactor tube, which can cause catalyst deactivation or blockage of the tubes [42]. Carbon growth will encapsulate and prevent access to active sites of nickel particles and thus reduce the activity of the catalyst [43]. According to the published researches, nano catalysts of core–shell sturcture prevent the formation or expansion of coke [33, 34, 44]. In this study, we intend to combine the advantages of Ni core with MCM-41 silica shell to develop a novel core–shell structure of NiO@MCM-41 nanocomposite by using vesicles as soft template. The developed nanocomposite, which to authors’ best knowledge is reported for the first time, can be used as a catalyst of reforming process. The main problem of methane reforming with carbon dioxide is the rapid deactivation of the nickel based catalyst. MCM-41 silica is a porous material with important features such as high surface area and porosity, uniform and adjustable pore size as well as high temperature resistance. The use of porous structure of MCM-41 as a thin shell, may protect the metal oxide core and improve resistance to coking by steric inhibition.
2 Experimental
2.1 Materials and preparation
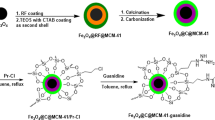
Ni(NO3)2·6H2O, Cetyl tri-methyl ammonium bromide (CTAB, 99%) and Sodium dodecyl benzene sulfonate (SDBS, 88%) were obtained from Sigma-Aldrich. Tetraethyl orthosilicate (TEOS) was obtained from Merck. The strategy for the formation of core–shell nanocomposite is shown in Fig. 1. First, 0.3 g CTAB was added to the solution of 144 ml of deionized H2O and 1.05 ml of NaOH (2 M) under stirring at 80 °C. After that, 1.5 ml of TEOS was added to the mixture. Stirring was continued for 1 h at 80 °C.

Schematic diagram of the formation mechanism of the NiO@MCM-41 nanocomposite
Vesicles were prepared by mixing CTAB and SDBS at the total concentration of 0.03 (mol L−1) and mixing mole ratio of 1.0:2.0 for 12 h at 30 °C. After that 0.25 g of Ni(NO3)2·6H2O was added to 50 mL of 0.03 (mol L−1) CTAB/SDBS (mole ratio of 1.0:2.0) solution which had been kept stagnant for 12 h. After 30 min of mild shaking, the pH value of the solution was adjusted to 8.5 by adding NaOH (0.01mol L−1). The resulting mixture was laid aside at room temperature in the dark for 3 h. Then the mixture was added to the MCM-41 gel with ultrasonic shaking for 60 min. The filtered precipitate was separated, washed several times with distilled water and ethanol, dried in oven at 50 °C, and calcinated at 550 °C for 10 h to obtain NiO@MCM-41 product.
2.2 Catalyst characterization
2.2.1 Low angle X-ray differaction measurement
Low and normal angle XRD spectra of synthesized core–shell nanocomposite samples was recorded by the diffractometer Philips PW1730 that uses the Cu Kα radiation with the power of 1200 watts (40 keV) and a wavelength of 1.54 Å to perform analysis with the 2θ scanning from 0.7 to 10 for low angel mode and 6–90 for normal angel.
2.2.2 Surface area analysis
The surface areas of the catalyst were determined in the − 196 °C by the micromeritics ASAP 2020 system. The porosity and specific surface were calculated based on the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) theory. The device to determine the surface area and porosity measures the absorption and desorption of nitrogen gas by the sample at a constant temperature of liquid nitrogen.
2.2.3 Field emission scanning electron microscopy (FESEM)
FESEM images and EDX spectrum were used to verify the formation and quantity of carbon structure. SEM images were obtained by TESCAN MIRA3 instrument with the accelerating voltage of 15 kV.
2.2.4 Transmission electron microscopy (TEM)
TEM images was used to verify the core–shell structure and to confirm the formation of mesoporous MCM-41. TEM images were obtained by Philips CM-10 instrument with the accelerating voltage of 200 kV.
2.2.5 Temperature programmed reduction (TPR)
The temperature programmed reduction analysis was performed by using Micrometrics Chemisorb 2750 instrument. In TPR analysis, a determined amount of the degassed calcined sample was heated under a reducing gas stream (10%H2 in Ar, 30 mL/min) with a temperature ramp of 10 °C/min. The amount of H2 uptake was monitored by the TCD detector.
2.2.6 Catalytic methane dry reforming reaction
CRM reaction was conducted in a fixed bed quartz reactor with an inner diameter of 5 mm and outer diameter of 7 mm. The experimental setup was shown in Fig. 2. In a typical experiment, the catalyst powder was mixed with bentonite with 1:1 weight ratio using some drops of water to obtain a homogenous paste. Then, the past of catalyst dried in oven at 90 °C for 4 h and calcined in an electric furnace, heating at 2.5 C/min to 550 °C and remained for 5 h. The diluted catalyst was pressed and crushed to the particles of 1 × 1.5 mm2. 0.15 gr of the catalyst with were placed inside the quartz reactor. The catalyst was heated to 500 °C with a ramp of 5 °C/min under atmospheric stream of Argon with the flow rate of 20 ml/min. After that, the catalyst was reduced for 2 h at 500 °C with stream of mixed H2 (10 ml/min) and Argon with molar ratio of H2:Ar = 1:4. Reaction was carried out at 850 °C with the gas feed flow rate (GHSV) of 48,000 ml h−1 gr active cat−1 and molar ratio of CH4:CO2 = 1:1 in the feed stream. The flow rate either of reactants was 30 ml/min. The exhaust gases, after passing through a vapor trap, were analyzed by gas chromatograph Agilent 7890 equipped with a TCD detector and thin film HP-PLOT MoleSieve capillary column of 30 m in length and 0.32 mm of inner diameter under He flow as the carrier gas. In each GC analysis of product samples, the oven temperature was kept isothermal at 35 °C, the injection temperature was at 120 °C, and the temperature of TCD detector was set to 150 °C.

Schematic of the laboratory reactor system
3 Results and discussion
3.1 XRD results
Low angle XRD analysis of nanocomposite sample was used to confirm the structure of regular mesoporous MCM-41 that was intended to be formed as the external shell of nano particles. As can be seen in Fig. 3a, the formation of three peaks at 2θ range of 2–6 indicated that the meso phase of the MCM-41 silica was formed as the shell for NiO@MCM-41 core–shell nanocomposite. Low angle X-ray diffraction pattern of the synthesized sample show three diffraction peaks of the crystallographic planes (100), (110) and (200) at 2θ equals 2 (d100 = 44.1 Å), 3.5 (a0 = d110 = 25.5 Å), and 4.2 (d200 = 21 Å), respectively. It represents the formation of the pores network with hexagonal structure in the shell region of nanocomposite particles. Reflection of plane (100) was broad and those of (110) and (200) were appeared quite wide with low intensity. Overspread and reducing the peak intensity can be due to the presence of the nickel oxide core in the nanocomposite with a much more dense crystal structure.

a Small angle XRD pattern of nanocomposite NiO@MCM-41, Fresh and coked catalysts, b Wide angle X ray diffraction pattern of the nanocomposite and the used catalyst. Diffraction data of inset figure were recorded in a different analysis
Low angel XRD spectra of fresh and spent catalyst did not show any diffraction pattern. These samples were obtained after calcining the dried paste of nanocomposite and Bentonite. The intensity of the X-ray beam will be reduced to almost zero after traveling through the specimen for a certain distance. Therefore, the diffracted beam intensity from below such a certain depth will be negligible and the information contained in the diffracted beam will only be due to the material above this depth [45]. Since the penetration depth of low-energy X-rays (< 50 keV) is shallow, traditional X-ray diffraction is thus unable to probe the interior of thicker structures. Others have used higher energies (> 150 keV) to study thicker samples [46]. Considering the above matter, the loss of diffraction peaks of hexagonal order of silica MCM-41 in the low angle xrd spectra of the mixed samples may be caused by the insufficient energy of the applied X ray (40 keV) especially at the low angel of the incident beam to the sample. It means that nanocomposite particles are located and covered in the matrix of bentonite.
Normal angle XRD analysis of samples before mixing with bentonite and after reaction was obtained to evaluate the present crystalline phases in the nanocomposite (before mixing with bentonite) and used catalyst (mixed with bentonite and tested in the reaction). Three groups of reflection of different crystalline phases were identified. As can be seen in Fig. 3b, the first group of broad reflections at 2θ ≈ 6–10, 15–30, 30–45 was attributed to (semi)crystalline phase of SiO2 in the pore walls of MCM-41 structure of shell part of nanocomposite [47]. The first reflection, 2θ ≈ 6–10, was present in both samples, whereas the other two reflections were disappeared in the used (coked) sample. The growth of coked in the shell part of nanoparticles may result in the crystal defects of SiO2 lattice and the disappearing of the latter reflections.
The second and third groups of reflections belong to the NiO and Ni crystal phases in the core of Nanoparticles. NiO lattice reflection appeared at 2θ = 38, 43.8, 63.2, 76.2, 79.8 [34, 48] and FCC Ni lattice reflection appeared at 2θ = 45, 52.2, 76.7 [35]. The absence and presence of reflection of metallic nickel in the spent catalyst proved the reduction of NiO to Ni in the core of nanparticles. The presence of distinct reflections of metallic Nickel lattice of spent sample indicated the stable condition of the metallic phase of core region of nano particles after the reaction. Presence of the graphitic structure of carbon formed on the spent catalyst was confirmed from the reflection at 2θ = 27.2 [35]. A reflection at 2θ ≈ 60, 61 in the xrd pattern of samples in Fig. 3b may be assigned to the nickel phyllosilicates phases [49], slightly weakening in the XRD pattern of the spent catalyst due to the reduction process.
3.2 N2 adsorption/desorption analysis
According to N2 adsorption/desorption isotherms of nanocomposite sample, given in Fig. 4a, it is observed that the absorption and desorption diagrams based on the IUPAC classification is placed in the category IV which is related to mesoporous solids. A hysteresis loop, formed from the branches of adsorption and desorption isotherms, is almost of H1 type. It is characteristic of hexagonally ordered mesoporous structure of MCM-41 particles. The inclined shape of hysteresis loop was reflected as two distinct bans in the BJH (Fig. 4b) pore size distribution curve. The two pore size bands, related to the two separated hysteresis loops, were supposed to be originated from the particles with and without NiO content in the core region. Particles with the larger NiO core lead to the larger pore sizes in their shell part, respectively.

Texture properties of nanocomposite, fresh catalyst, and coked catalyst: N2 adsorption and desorption isotherms; left, right. Distribution of pore size obtained from desorption branch of N2 isotherms. Fresh and coked catalysts were degassed at 350 and 150 °C, respectively
Considering height of hysteresis loop, the second deconvoluted hysteresis loop represented a higher capacity of N2 adsorption. It means the major portion of composite particles comprised of the core of nickel oxide.
The N2 isotherm data of fresh catalyst showed the similar shape to that of the nanocomposite sample. The mixing the nanocomposite with the bentonite with lower surface area resulted in the deceased surface area and pore volume. The meso-porosity of the parent nanocomposite was remained in the mixed sample as the isotherm data showed the hysteresis loop started at P/P0 ≈ 0.45. Therefore, the presence of Hexagonal order of MCM-41 pore structure can be verified in the fresh catalyst, which is in contrary to the low angel XRD spectrum. The N2 isotherm data of the coked catalyst represented a substantial decrease of the specific surface area which is an indication of carbon formation in the partial mesoporous structure. It should be noted that the coked sample was degassed at 150 °C to ensure preservation of carbon species from probable pyrolysis at higher temperatures. The rather low degas temperature may lead to measurement of a lower adsorption capacity for the coked sample.
According to Fig. 4b, pore size distribution diagram represented two bands. The first narrow and intense band of the smaller pores was related to the particles without nickel core. The second wide and short band of the larger pores was assigned to the particles with smaller core size, leading to the steeper part of the hysteresis loop in Fig. 4a. The texture features of samples are summarized in Table 1. It can be seen that the synthesized nanocomposite has a reduced surface area (617 m2 g−1) compared to pure MCM-41 silica. BJH model predicts two meso-pore sizes of 7.4 and 3.9 nm. It should be pointed out that the larger calculated size of mesopores of the prepared nanocomposite is about twofold of the typical values (3–4 nm) reported with the common synthesis method of silica MCM-41. This is a nonphysical pore size of BJH model, resulted due to core–shell structure of particles. The nonporous core part of nanocrystallites located inside of the porous structure of MCM-41, leading to reduced adsorption capacity and surface area of the whole particle compared to a pure MCM-41 particle of the same size and mass. Therefore, the reduced measured adsorbed N2 gas per gram of sample resulted in the smaller surface area and the larger pore size compared to MCM-41 silica. However, the real pore size of mesoporous shell is less in the range of 1.5–2 nm as can be calculated from the results of low angel XRD and TEM analyses.
Mixing of the nanocomposite with bentonite followed by calcination at 550 °C resulted in the reduction of pores volume, as can be seen in Fig. 4a and Table 1. One reason may be the solid state reaction of the porous structure of MCM-41 particles at their borders with the bentonite particles and the subsequent partial blockage of the porous structure of silica MCM-41. According to Fig. 4b, the decrease of the larger pore size of the fresh catalyst compared to the nanocomposite may be explained by the probable surface diffusion and deposition of bentonite crystallites into these pores.
After dry reforming reaction at 850 °C, the fresh catalyst encountered the severe loss of pores volume due to the formation of carbonaceous species in the pore structure of the sample. As shown in the Fig. 4b, the previous mesopores in the BJH distribution of pores size of parent samples disappeared.
3.3 TEM
Figure 5 shows the TEM images of NiO@MCM-41 nanocomposite. The most of nanocrystallites indicated a core–shell structure with the minor homogenous nanocrystallites of MCM-41. As could be seen in Fig. 5, the particle size was about 70–80 nm with narrow distribution and the minimum shell thickness was about 15–20 nm, although, particles of small and large metal oxide core and no core inside were present, simultanously. Image of part (b) was enlarged and was given in the part (c), where hexagonal structure of pores of MCM-41 domain around the metal oxide core marked with the red hexagons for identification. It confirmed the formation of MCM-41 mesoporous silica in the shell region of nanocomosite and homogenous nanocrystallites with no core region. Using image processor tools for quantified analysis of TEM images, if wall thickness of pores is 1 nm, the estimated size of meso-pores would be 2 nm. It is well comparable to the pore size of ca. 1.5 nm (a0-1) obtained from XRD data by using Bragg law (Fig. 6).

TEM images of NiO@MCM-41 nano crystals, a zoomed out, b zoomed in, c image (b) was enlarged, cropped and recombined and the hexagonal order of pores was made distinguishable via red polygons. (Color figure online)

Temperate programmed reduction profile of the fresh catalyst
3.4 Temperature programmed reduction
Ni/SiO2 catalyst prepared by impregnation usually has a major hydrogen consumption peak at ca. 430 °C and small hydrogen consumption peak around 500 °C [50,51,52,53]. The first reduction peak (430 °C) is usually attributed to the reduction of NiO, with a weak interaction with the silica support and the second peak (500 °C) is attributed to NiO with stronger interaction with the support. Shift of the reduction temperature of nickel species to higher temperature [54, 55] indicates greater integration between NiO and the oxide support. This interaction can enhance resistance for coke deposition and prevent sintering of nickel particles [49]. According to the literature [50, 56,57,58,59] during deposition–precipitation of nickel with silica spheres, usually only nickel hydroxide and nickel phyllosilicate is formed, although formation of nickel(II) basic carbonate is also possible [60]. By decomposition of urea during nickel deposition–precipitation nickel hydroxide formed from nickel salt reacts with silicic acid to form Ni–O–Si bonds [59]. With the formation of Ni–O–Si bond, two forms of nickel silicates have been verified from nickel hydroxide: 1:1 phyllosilicate Ni3(Si2O5)(OH)4 and 2:1 phyllosilicate Ni3(Si2O5)2(OH)2 [57, 58].
Burattin et al. [57, 61,62,63] reported that 1:1 nickel phyllosilicate is reduced at temperatures in the range 450–650 °C on Ni/SiO2 catalysts, whereas 2:1 nickel phyllosilicate requires higher temperatures in the range 690–760 °C. Reduction of the fresh catalyst started beyond 350 °C and the maximum hydrogen consumption was at 700 °C. Two minor reduction peaks were obtained at 412 and 510 °C. The appearance of three separate peaks during catalyst reduction suggests the occurrence of different Ni phases on the catalyst surface. The TPR profile of fresh catalyst shows two distinct peaks in the range typical for phyllosilicate and thus it can be concluded that both forms of nickel phyllosilicate are present. According to TPR results of Majewski et al. [49], the hydrogen consumption above 440 °C with the maximum reduction peak at 700 °C can be attributed to the reduction of 2:1 nickel phyllosilicate. Part of the minor hydrogen adsorption peak at 512 °C can be also attributed to 1:1 nickel phyllosilicate. The small hydrogen consumption at 410 °C observed by TPR can be attributed to unsupported nickel hydroxide [61] or possibly NiO crystallites without support interaction. Major hydrogen consumption at 700 °C for the prepared catalyst is a significant achievement of this work.
3.5 Catalytic performance
Results of catalytic performance of sample in the dry reforming of CH4 to synthesis gas were shown in Fig. 7. According to Fig. 7a, the conversion of reaction increases over time, indicating that the intermediate concentration formed on the catalyst surface increases during the first 245 min of time on stream. The conversion efficiency of CH4 and CO2 after 245 min of reaction stabilized and rised to 42 and 48%, respectively. The CO yield, as given in Fig. 7b, increases from 5 to around 25 wt% at the same period of time.

a CH4 and CO2 conversions, b CO and H2 yields, c ratio of yield of H2 to CO with respect to the reaction time at 850 °C, CH4:CO2 = 1, CH4 flow = 30 ml/min (GHSV = 48,000 ml h−1 gr active cat−1), 1 atm
In the most recent studied catalyst of similar species but with different structure reported by Zhang et al. [48], the catalyst composed of Ni clusters dispersed in MCM-41 porous structure showed CH4 and CO2 conversion of ca. 95 and 97% at 800 °C and GHSV = 22,500 ml h−1 gr cat−1, respectively. The reactor experiment was carried out at less than half of the GHSV used in this study.
In another study by Zheng et al. [34], the catalytic performance of LaNiO3@SiO2 core–shell nano-particles were evaluated in CRM by a coaxial dielectric barrier discharge (DBD) plasma reactor at GHSV = 12,000 ml h −1 gr cat−1. The temperature of catalysts bed was measured 200 °C. The synergistic CH4 and CO2 conversion were ca. 87 and 78%, where the share of the catalysts in either the conversions of species were at least about 35%.
Zhang et al. [35] were used Ni@SiO2 catalyst for CRM at the temperature of 750 °C and feed flow rate of GHSV = 48,000 cm3 h−1 gr cat−1. CH4 and CO2 conversion of ca. 60 and 70% were obtained with slow deactivation rate, respectively. But, they achieved to yH2/yCO ratio of about 0.7 after 5 h reaction.
Kim et al. [33] synthesised Ni@TiO2 catalyst and studied its performance in CRM at T = 800 °C, undiluted feed flow of 20 ml/min with H2:CO molar ratio of 1:1. At the initial stage of the CRM reaction at 800 °C, all of the catalysts showed high catalytic activities with CH4 and CO2 conversions of approximately 50–59 and 56–58%, respectively. The bare Ni catalyst showed the highest initial conversions and Ni@TiO2 with the thicker shell led to the lowest value. However, the deactivation rate by coke deposition was reversed with respect to the catalysts. At the average, during 169 h of the CRM reaction at 800 °C, all of the catalysts showed CH4 and CO2 conversions of approximately 32.5–38 and 48–50%, respectively.
After 245 min of time on stream the reaction, the ratio of production yield of H2:CO approached to the values of less than one (Fig. 7c).
The following reactions (1–3) including steam reforming, dry reforming, and reverse water gas shift can compete with each other in the course of CRM conversion at the temperature of 850 °C which is much higher than temperature 707 °C required for their thermal activation.
With consideration of the decreasing trend of H2/CO ratio with time, the enhancement of reverse water gas shift reaction with time on stream can be deduced to vary the H2/CO ratio in the favour of CO. According to the results of the catalytic performance, as H2 and CO concentration rises with time, the reverse water gas shift reaction strengthen due to H2 build-up (or adsorbed atomic H) near to metal surface.
According to results, NiO@MCM-41 core–shell nano composite showed a delayed final conversion. This observation may be explanied with two possible reasons. First, the limiting effect of shell of MCM-41 silica on the intra-particle diffusion of the reacting species to the metal core may reduce the rate of coverage of metalic surface. Furthermore, one dimensional pore structure of MCM-41 as the shell layer could intensify the barrier effect on the diffusion of species to the active core. The other contributing phenomenon may be the progressive reduction of remaining nickel oxides with time on stream at the CRM conditions. TPR data showed temperature as high as 700 °C is required for the complete reduction of the prepared catalyst. Methane as reactant, H2, and CO as products of reaction can reduce the nickel oxide left from incomplete pre-reduction step. Based on the TPR result duration of about 30 min is estimated for complete reduction of catalyst exposed to reactant stream and products at 850 °C.
Possibly, the dilution of the catalyst and its active sites with bentonite not only resulted in a rather long delay in reaching to the final activity of the catalyst but also it reduced the maximum activity of catalyst compared to the above reported similar researches. It should be noted that manufacturing process of industrial catalysts passes a shaping step using binders which is usually not considered in the laboratory researches of catalytic reactions.
Regarding the enhanced sintering and coke resistance of previously reported core–shell Ni catalysts [38,39,40], the prepared catalyst can potentially be applied for CRM and other high temperature hydrocarbon conversions.
It needs longer activity time to determine the influence of MCM-41 shell on the life time of the prepared catalyst, as well as its resistance against the coke formation and sintering of the metal core in the high temperature of CRM reaction.
In addition to the diffusion barrier to species transport, the formation of a thick shell region around the active core of catalyst nanocrystals can adversely affect the heat transport to the endothermic CRM reaction. Therefore, the study of effect of core diameter and shell thickness of the nanocomposite in the CRM reaction can improve their catalytic performance.
3.6 SEM/EDX analysis of the spent catalyst
The deactivation of the Ni-based catalyst during the CRM reaction is a well-known phenomenon due to the coke formation on the active sites of the catalyst by the CH4 cracking and the reverse-Boudouard [33]. The EDX data, given in Table 2, showed that carbonaceous specious were formed after the catalytic CRM reaction at 850 °C for duration of 4 h. Based on the EDX data of Ni contents and assumption of Ni dispersion = 100%, TOF of methane conversion in CRM of the catalyst was approximatedly reached to 0.69 s−1 after 4 h of CRM reaction. The average rate of carbonaceous specious formation can be similarly approximated as TOF = 8 × 10−4 s−1.
SEM images of the spent catalyst were given in Fig. 8. It seems that particles of spent catalyst became severely agglomerated and formed a large continuum domain of micrometers thickness under high reaction temperature of 850 °C. The exposed surface of the continuum domain of catalyst seems to be covered and smoothen by carbon layer. The growth of carbonaceous carbon around some isolated particles of smaller size probably has resulted in the formation of filament carbon which can be seen in the center of the first image of the first row and zoomed in the second image of the first row. The image of an agglomerated particle observed in the first image of the second row has been magnified and shown in the second image of the same row. The measured nano-metric sizes of nanoparticles have been marked on the latter image, which every size is about 20 nm. It means that sintering of nickel core in the reaction condition of high temperature has been prevented by the protecting layer of MCM-41 as nanparticles could be observed in the SEM images.

SEM images of the spent catalyst at two magnification
4 Conclusions
We have demonstrated a two-step soft templated nanocasting method to synthesize NiO@MCM-41 nanocomposite with a core/shell structure. The results of the N2 adsorption and desorption and XRD analyses confirmed the formation of MCM-41 in the nanocomposite. The results of the TEM confirmed the formation of core–shell structure with mesoporous MCM-41 silica shell covered the nickel oxide core. The size of cores was determined by vesicles, where formed with anionic/cationic surfactant self-assembly in aqueous solution. The average particle size of core/shell structure is in the range 70–80 nm with the minimum thickness of porous shell layer of 15–20 nm. Particles formed as composite of either small or large metal oxide core while part of particles showed no core inside. After 245 min of time on stream, the catalytic activity was nearly stabilized and the conversion efficiency of CH4 and CO2 with the prepared catalyst of Ni@MCM-41 reached to 42 and 48%, respectively. Catalyst TOF of methane conversion was at least 0.69 s−1 after 4 h of CRM reaction. The core–shell nanocomposite of Ni@MCM-41 catalyzed the CRM with H2/CO yield ratio of ca. 0.8. The prepared nanocomposite presented potentials for CRM and other high temperature hydrocarbon conversions. Hydrogen consumption at 700 °C by the prepared catalyst showed the strong interaction of the major Nickel content with the support. SEM analysis provided evidence of stability of the prepared catalyst against sintering. The optimization of the core diameter and shell thickness of the nanocomposite may improve the catalytic performance in the CRM reaction.
References
B.L. Cushing, V.L. Kolesnichenko, C.J. O’Connor, Recent advances in the liquid-phase syntheses of inorganic nanoparticles. Chem. Rev. 104, 3893–3946 (2004)
R. Davies et al. Engineered particle surfaces. Adv. Mater. 10(15), 1264–1270 (1998)
C. Hofman-Caris, Polymers at the surface of oxide nanoparticles. New J. Chem. 18(10), 1087–1096 (1994)
L.M. Liz-Marzán, M. Giersig, P. Mulvaney, Synthesis of nanosized gold-silica core–shell particles. Langmuir 12(18), 4329–4335 (1996)
R. Partch, In Material Synthesis and Characterization, D. Perry (ed.) (Plenum, New York, 1997)
D. Wilcox et al., Hollow and Solid Spheres and Microspheres. (Materials Research Society, Pittsburg, 1995)
D.V. Goia, E. Matijević, Preparation of monodispersed metal particles. New J. Chem. 22(11), 1203–1215 (1998)
M. Ohmori, E. Matijević, Preparation and properties of uniform coated inorganic colloidal particles: 8. Silica on iron. J. Colloid Interface Sci. 160(2), 288–292 (1993)
R. Partch, S. Brown, Aerosol and solution modification of particle-polymer interfaces. J. Adhes. 67(1–4), 259–276 (1998)
F. Wang, L. Xu, W. Shi, Syngas production from CO2 reforming with methane over core-shell Ni@ SiO2 catalysts. J CO2 Utilization 16, 318–327 (2016)
S.-J. Oh et al., Newly designed Cu/Cu10Sn3 core/shell nanoparticles for liquid phase-photonic sintered copper electrodes: large-area, low-cost transparent flexible electronics. Chem. Mater. 28(13), 4714–4723 (2016)
Z. Zhou et al., Preparation of calcium carbonate@graphene oxide core–shell microspheres in ethylene glycol for drug delivery. Ceram. Int. 42(2), 2281–2288 (2016)
L.E. Sperling et al., Advantages and challenges offered by biofunctional core–shell fiber systems for tissue engineering and drug delivery. Drug Discov Today 21(8), 1243–1256 (2016)
Z. Shen et al., Self-assembly of core-polyethylene glycol-lipid shell (CPLS) nanoparticles and their potential as drug delivery vehicles. Nanoscale 8(31), 14821–14835 (2016)
D. Saikia et al., Aqueous synthesis of highly stable CdTe/ZnS Core/Shell quantum dots for bioimaging. Luminescence 32(3), 401–408 (2016)
L. Tan et al., Chitosan-based core-shell nanomaterials for pH-triggered release of anticancer drug and near-infrared bioimaging. Carbohydr. Polym. 157, 325–334 (2017)
R.A. Perez, H.-W. Kim, Core–shell designed scaffolds for drug delivery and tissue engineering. Acta Biomater 21, 2–19 (2015)
E. Yan et al., Degradable polyvinyl alcohol/poly (butylene carbonate) core–shell nanofibers for chemotherapy and tissue engineering. Mater. Lett. 167, 13–17 (2016)
H.S.A. El-Haleem et al., Manganese dioxide-core–shell hyperbranched chitosan (MnO2–HBCs) nano-structured screen printed electrode for enzymatic glucose biosensors. RSC Adv. 6(110), 109185–109191 (2016)
C. Zhang et al., Electrochemical biosensor based on nanoporous Au/CoO core–shell material with synergistic catalysis. ChemPhysChem. 17(1), 98–104 (2016)
R. Zeng et al., Aqueous synthesis of type-II CdTe/CdSe core–shell quantum dots for fluorescent probe labeling tumor cells. Nanotechnology 20(9), 095102 (2009)
A. Haghtalab, A. Mosayebi, Co@ Ru nanoparticle with core–shell structure supported over γ-Al2O3 for Fischer–Tropsch synthesis. Int. J. Hydrogen Energy 39(33), 18882–18893 (2014)
A. Shafiefarhood et al., Fe2O3@ LaxSr1 –xFeO3 core–shell redox catalyst for methane partial oxidation. ChemCatChem 6(3), 790–799 (2014)
J.-X. Feng et al., Caffeine-assisted facile synthesis of platinum@ palladium core-shell nanoparticles supported on reduced graphene oxide with enhanced electrocatalytic activity for methanol oxidation. Electrochim. Acta 142, 343–350 (2014)
M. Zhang et al., Improved catalytic activity of cobalt core–platinum shell nanoparticles supported on surface functionalized graphene for methanol electro-oxidation. Electrochim. Acta 158, 81–88 (2015)
M.A. Lucchini et al., Sintering and coking resistant core–shell microporous silica–nickel nanoparticles for CO methanation: towards advanced catalysts production. Appl. Catal. B 182, 94–101 (2016)
W. Qi et al., Core–shell nanostructures: modeling, fabrication, properties, and applications. J. Nanomaterials 2012, 2 (2012)
J. Hao et al., Zn2+-induced vesicle formation. J. Phys. Chem. B 108(4), 1168–1172 (2004)
W. Song et al., Synthesis of magnetic core–shell structure Fe3O4@ MCM-41 nanoparticle by vesicles in aqueous solutions. Chin. J. Chem. Eng. 23(8), 1398–1402 (2015)
N. Zhao et al., Controlled synthesis of gold nanobelts and nanocombs in aqueous mixed surfactant solutions. Langmuir 24(3), 991–998 (2008)
L. Fan, R. Guo, Growth of dendritic silver crystals in CTAB/SDBS mixed-surfactant solutions. Cryst. Growth Des. 8(7), 2150–2156 (2008)
F. Liu et al., Transcriptive synthesis of Mg(OH)2 hollow nanospheres and the non-equilibrium shell fusion assisted by catanionic vesicles. J. Phys. Chemis. B 113(33), 11362–11366 (2009)
D.H. Kim et al., The catalytic stability of TiO2-shell/Ni-core catalysts for CO2 reforming of CH4. Appl. Catal. A 495, 184–191 (2015)
X. Zheng et al., LaNiO3@ SiO2 core–shell nano-particles for the dry reforming of CH4 in the dielectric barrier discharge plasma. Int. J. Hydrogen Energy 39(22), 11360–11367 (2014)
J. Zhang, F. Li, Coke-resistant Ni@ SiO2 catalyst for dry reforming of methane. Appl. Catal. B 176, 513–521 (2015)
E. Baktash et al., Alumina coated nickel nanoparticles as a highly active catalyst for dry reforming of methane. Appl. Catal. B 179, 122–127 (2015)
P. Ferreira-Aparicio, A. Guerrero-Ruiz, I. Rodrıguez-Ramos, Comparative study at low and medium reaction temperatures of syngas production by methane reforming with carbon dioxide over silica and alumina supported catalysts. Appl. Catal. A 170(1), 177–187 (1998)
J. Edwards, A. Maitra, The chemistry of methane reforming with carbon dioxide and its current and potential applications. Fuel Process. Technol. 42(2–3), 269–289 (1995)
M. Bradford, M. Vannice, CO2 reforming of CH4. Catal. Rev. 41(1), 1–42 (1999)
J. Rostrupnielsen, J.B. Hansen, CO2-reforming of methane over transition metals. J. Catal. 144(1), 38–49 (1993)
X. Gao et al., Highly reactive Ni-Co/SiO2 bimetallic catalyst via complexation with oleylamine/oleic acid organic pair for dry reforming of methane. Catal. Today 281, 250–258 (2017)
J.R. Rostrup-Nielsen, Industrial relevance of coking. Catal. Today 37(3), 225–232 (1997)
H. Swaan et al., Deactivation of supported nickel catalysts during the reforming of methane by carbon dioxide. Catal. Today 21(2), 571–578 (1994)
L. Neal, A. Shafiefarhood, F. Li, Effect of core and shell compositions on MeOx@ LaySr1–yFeO3 core–shell redox catalysts for chemical looping reforming of methane. Appl. Energy 157, 391–398 (2015)
J. Liu, R.E. Saw, Y.-H. Kiang, Calculation of effective penetration depth in X-ray diffraction for pharmaceutical solids. J. Pharm. Sci. 99(9), 3807–3814 (2010)
D.W. Fitting, W.P. Dube, T.A. Siewert, High energy x-ray diffraction technique for monitoring solidification of single crystal castings. Nondestruct. Charact. Mater. 8, 217–222 (1998)
M. OOKAWA et al., X-ray diffraction analysis of ordered mesoporous silica. Stud. Surf. Sci. Catal. 135(1), 198–198 (2001)
Q. Zhang et al., A sintering and carbon-resistant Ni-SBA-15 catalyst prepared by solid-state grinding method for dry reforming of methane. J. CO2 Utilization 17, 10–19 (2017)
A.J. Majewski, J. Wood, W. Bujalski, Nickel–silica core@shell catalyst for methane reforming. Int. J. Hydrogen Energy 38(34), 14531–14541 (2013)
A. Loaiza-Gil et al., Thermal decomposition study of silica-supported nickel catalyst synthesized by the ammonia method. J. Mol. Catal. A: Chem. 281(1), 207–213 (2008)
Y. Matsumura, T. Nakamori, Steam reforming of methane over nickel catalysts at low reaction temperature. Appl. Catal., A 258(1), 107–114 (2004)
Y.-X. Pan, C.-J. Liu, P. Shi, Preparation and characterization of coke resistant Ni/SiO2 catalyst for carbon dioxide reforming of methane. J. Power Sour. 176(1), 46–53 (2008)
H.-W. Chen et al., Carbon dioxide reforming of methane reaction catalyzed by stable nickel copper catalysts. Catal. Today 97(2), 173–180 (2004)
A. Djaidja et al., Characterization and activity in dry reforming of methane on NiMg/Al and Ni/MgO catalysts. Catal. Today 113(3), 194–200 (2006)
L.-j. SI et al., Influence of preparation conditions on the performance of Ni–CaO–ZrO2 catalysts in the tri-reforming of methane. J. Fuel Chem. Technol. 40(2), 210–215 (2012)
P. Jin et al., Synthesis and catalytic properties of nickel–silica composite hollow nanospheres. J. Phys. Chem. B 108(20), 6311–6314 (2004)
P. Burattin, M. Che, C. Louis, Characterization of the Ni(II) phase formed on silica upon deposition–precipitation. J. Phys. Chem. B 101(36), 7060–7074 (1997)
T. Lehmann et al., Physico-chemical characterization of Ni/MCM-41 synthesized by a template ion exchange approach. Microporous Mesoporous Mater. 151, 113–125 (2012)
J.C. Park et al., Chemical transformation and morphology change of nickel–silica hybrid nanostructures via nickel phyllosilicates. Chem. Commun. 2009(47), 7345–7347
M. Ocana, A. Gonzalez-Elipe, Preparation and characterization of uniform spherical silica particles coated with Ni and Co compounds. Colloids Surf. A 157(1), 315–324 (1999)
P. Burattin, M. Che, C. Louis, Molecular approach to the mechanism of deposition–precipitation of the Ni(II) phase on silica. J. Phys. Chem. B 102(15), 2722–2732 (1998)
P. Burattin, M. Che, C. Louis, Metal particle size in Ni/SiO2 materials prepared by deposition–precipitation: Influence of the nature of the Ni(II) phase and of its interaction with the support. J. Phys. Chem. B 103(30), 6171–6178 (1999)
P. Burattin, M. Che, C. Louis, Ni/SiO2 materials prepared by deposition–precipitation: influence of the reduction conditions and mechanism of formation of metal particles. J. Phys. Chem. B 104(45), 10482–10489 (2000)
Acknowledgements
Authors gratefully acknowledge the financial support of Iran Nanotechnology Initiative Council (INIC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Roosta, Z., Izadbakhsh, A., Sanati, A.M. et al. Synthesis and evaluation of NiO@MCM-41 core–shell nanocomposite in the CO2 reforming of methane. J Porous Mater 25, 1135–1145 (2018). https://doi.org/10.1007/s10934-017-0525-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-017-0525-8