
Abstract
Multifunctional carbon aerogel was prepared by pyrolysis of sodium carboxymethyl cellulose aerogel after sol–gel processing and freeze-drying. Ferric iron and D-(+)-gluconic acid-lactone were used as cross-linker and releasing agent, respectively. The as-prepared carbon aerogel was characterized by scanning electron microscopy, Fourier transform infrared spectroscopy, X-ray diffractometry, thermogravimetric analysis, nitrogen adsorption measurements, vibrating sample magnetometry and water contact angle tests. The carbon aerogel had a high specific surface area of 742.34 m2/g and contained mostly mesopores. Its density was as low as 0.062 g/cm3. The carbon aerogel morphology has an uneven three-dimensional structure and presented a type-IV adsorption isotherm. Carbonization resulted in the destruction of the carboxymethyl cellulose crystalline structure, damage to the oxygen-containing functional groups and provided the carbon aerogel with good magnetic properties. The carbon aerogel displayed good hydrophobicity and flame retardance, which has potential application in adsorption, hydrophobic materials and fireproofing.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Carbon aerogel is a novel carbonaceous material with numerous excellent properties, including high specific surface, rich porosity, low density and high electrical conductivity [1–3]. Carbon aerogel has potential for application in many areas, such as catalyst supports, adsorbents, hydrogen storage, electrodes and supercapacitors [4–6]. Conventionally, carbon aerogel is obtained from carbonization of organic aerogel. Resorcinol and formaldehyde are used as raw materials to synthesize organic aerogel [7]. However, this synthesis method is time-consuming and costly supercritical drying is required. Furthermore, resorcinol and formaldehyde are poisonous and harmful to public health and environmental safety, which hinders developments in carbon aerogel. Researchers are striving to find a simple and low-cost approach to carbon aerogel preparation. Carbon aerogel from natural polymers, such as regenerated cellulose, and bacterial celluloses have a good adsorption capacity, fire resistance, electrical conductivity and magnetic properties, which has attracted researcher attention [4, 5, 8, 9]. Cellulose derivatives such as carboxymethyl cellulose (CMC) could also be used to prepare carbon aerogels and have ideal prospects.
We have reported on a facile, low-cost and environmentally friendly method to prepare carbon aerogel by sol–gel, freeze-drying and pyrolysis using CMC as raw materials. The as-prepared carbon aerogel was characterized by scanning electron microscopy (SEM), X-ray energy dispersive spectrometer (EDS), Fourier transform infrared (FT-IR) spectroscopy, X-ray diffractometry (XRD), thermogravimetric analysis (TGA), nitrogen adsorption, vibrating sample magnetometry (VSM) and water contact angle (WCA) tests. As-prepared carbon aerogel was also tested for magnetism, hydrophobicity and flame retardance.
2 Experimental
2.1 Materials
Carboxymethyl cellulose sodium (CMC), ammonia solution (25 %), FeCl3·6H2O (analytical reagent) and D-(+)-gluconic acid-lactone were used as cross-linkers and releasing agents. All chemical reagents were of analytical grade and were used as received without further purification.
2.2 Preparation of carbon aerogel
The CMC aerogel was prepared according to a previous report by Lin [10]. CMC (1.5 g) was added to 50 ml deionized water followed by 0.882 g D-(+)-gluconic acid-lactone. The mixture was stirred for 1 h at room temperature until a transparent solution was obtained. FeCl3·6H2O (0.882 g) was dissolved in deionized water and ammonia solution was used to adjust the solution pH to ~ 7. This solution was mixed with the CMC solution and stirred for 15 min to ensure homogeneity. The mixed solution was poured into a Petri dish and gelation occurred. The gel was kept for 3 days at room temperature for further gelation and cross-linking. The hydrogel was flushed with deionized water several times. Subsequently, hydrogels underwent freeze drying at −50 °C for 60 h in a vacuum to obtain CMC aerogel. The as-prepared CMC aerogel was carbonized at 950 °C in a tube furnace for 2 h at 5 °C/min under nitrogen to obtain a carbon aerogel.
2.3 Characterization
The surface morphology and elemental compositions were observed and determined by scanning electron microscopy (SEM) (FEI, Quanta 200) and X-ray energy dispersive spectrometer (EDS) and samples were sputter-coated with a thin layer of gold before analysis. X-ray diffraction (XRD) patterns were obtained to characterize the crystal structure with Cu Kα radiation at 5°/min from 5° to 80° 2θ (Rigaku, D/max 2200). Fourier transform infrared spectroscopy (FT-IR) was used to analyze sample functional groups from 650 to 4000 cm−1 (Nicolet, MAGNA-IR560 E.S.P). Thermal stabilities were determined using a thermal gravimetric analyzer (TGA) (Q500). Pore parameters were measured from the nitrogen adsorption isotherm at −196 °C based on Brunauer–Emmet–Teller (BET) and Barrett–Joyner–Halenda methods (ASAP 2020). Magnetic properties were investigated by VSM from −10000 to 10000 Oe at room temperature (Quantum Design, PPMS-9). The hydrophobicity was calculated using the WCA tester (Dataphysics, OCA20).
3 Results and discussion
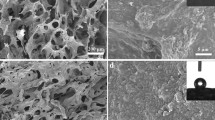
The CMC aerogel and carbon aerogel surface morphology are shown in Fig. 1. An intertwined and tight network structure was visible in the CMC aerogel. After carbonization, the carbon aerogel formed a three-dimensional structure. As shown in Fig. 1c that there existed packed, sheet-like microstructures which were separated by pores larger than 10 micrometers. While, large amounts of pores <50 nm were found on the surface of the sheet-like microstructure at high magnification in Fig. 1d, indicating that pyrolysis provided the carbon aerogel with numerous mesopores and an uneven and disorganized surface. Additionally, the oxygen content of carbon aerogel in EDS spectrum decreased significantly as shown in Fig. 1e compared with the CMC aerogel shown in Fig. 1b, which suggests that the oxygen-containing functional groups were mostly damaged during pyrolysis. Carbon aerogel density was measured from mass and volume measurements and was 0.062 g/cm3. Figure 1f shows that a chlorophytum leaf did not bend under the weight of three 1 cm × 1 cm × 0.5 cm carbon aerogels, which confirms its low density.

SEM image a and EDS spectrum b of CMC aerogel, SEM images at low magnification c, high magnification d and EDS spectrum of carbon aerogel e and photograph of carbon aerogels on chlorophytum leaf f
FT-IR spectra of CMC and CMC aerogel are shown in Fig. 2a. In CMC, two typical bands at 1600.37 and 1418.34 cm−1 occur because of carboxylate ion asymmetry stretching and symmetry vibration, respectively [10]. A broad and strong peak exists at 3295.66 cm−1, which is assigned to the CMC hydroxyl group. Adsorption peaks at 2918.70 and 1330.04 cm−1 are assigned to C–H stretching and bending vibrations [11]. Bends at 1016.25 cm−1 are attributed to sugar ring absorption [12]. The CMC aerogel spectrum was similar to that of CMC, but a new peak appeared at 1730 cm−1 from the addition of D-(+)-gluconic acid-lactone as reported in a previous study [13]. The adsorption band intensity at 2918.70 and 1330.04 cm−1 decreased, which indicates that the intermolecular hydrogen bands increased and that the interaction between CMC molecules strengthened [10].

FT-IR spectra of raw CMC and CMC aerogel a and XRD patterns of raw CMC, CMC aerogel and carbon aerogel b
XRD was used to investigate the crystal structure of raw CMC, CMC aerogel and carbon aerogel. As shown in Fig. 2b, the raw CMC exhibited two characteristic diffraction peaks at 2θ equal to 19.52° and 34.54°, which occurs because of the typical cellulose II crystal structure [14]. In the CMC aerogel, the characteristic CMC peaks nearly disappeared, which indicates that crosslinking occurred between CMC and Fe3+; the CMC molecules rearranged, which resulted in crystal structure damage. Several new peaks appeared at 22.77°, 32.49°, 46.60°, 52.55°, 58.21°, 68.43° and 77.98° from the generation of ammonium chloride crystals during CMC aerogel preparation [15]. After carbonization, the characteristic CMC peak disappeared and several new peaks emerged in the carbon aerogel. Peaks at 25.90° and 44.73° correlate with graphite planes, which is related to the formation of amorphous carbon [16–18]. Peaks at 29.73°, 33.56°, 43.17°, 54.05°, 57.7° and 61.13° correlate with a cubic Fe3O4 crystalline phase [19, 20], which illustrates that Fe3O4 was generated during pyrolysis.
CMC aerogel and carbon aerogel TGA and DTG curves (Fig. 3) were used to investigate the thermal stability. A two-stage mass loss occurred in the CMC aerogel. The first mass loss at 30–100 °C is ascribed to the evaporation of absorbed water [21]. The second significant mass loss occurred at 120–350 °C, and was caused by CMC decomposition [22]. The maximum decomposition rate of CMC aerogels was determined at 220 °C and the mass decreased by ~63.94 % in total. Compared with CMC aerogel, the carbon aerogel had only one obvious mass loss stage at 30–100 °C from remanent water in the products. The total mass loss was as low as 8.92 %, which proves that the carbon aerogel had good thermal stability.

TGA a and DTG b curves of CMC aerogel and carbon aerogel
Nitrogen adsorption was carried out to characterize the porous carbon aerogel structure. As shown in Fig. 4a, samples exhibited a representative type-IV adsorption isotherm and an obvious hysteresis loop at a relative pressure (P/P0) above 0.45 owing to capillary condensation according to the IUPAC classification [23]. This demonstrates the existence of mesopores. The specific surface area of the carbon aerogel was 742.34 m2/g obtained by the BET method. Furthermore, as shown in Fig. 4b, the pore diameters of the carbon aerogel varied from 2 to 75 nm, which indicates that the carbon aerogel was composed mainly of mesopores (2–50 nm) [4]. The large specific surface areas and high percentage of carbon aerogel mesopores ensured potential applications in the field of electrode materials because the pore size distribution played a significant role in chemical electrodes [7].

Nitrogen adsorption/desorption isotherms a and pore size distribution of carbon aerogel b
The magnetic properties of the carbon aerogel were characterized by VSM from −10000 to 10000 Oe. The magnetic hysteresis loop is S-shaped as shown in Fig. 5a, which indicates that the saturation magnetization, remanent magnetization and coercivity were 19.97, 0.53 emu/g and 67.75 Oe, respectively. The remanent magnetization was nearly zero, which demonstrates that when the external magnetic field is removed, no remaining magnetization existed [24]. Figure 5b shows that the carbon aerogel could be moved by a magnet, whereas the CMC aerogel could not be lifted under an external magnetic field as shown in Fig. 5c. The carbon aerogel has potential application such as in adsorption, ferrofluid technology and magnetocaloric refrigeration [25].

Magnetic hysteresis loop of carbon aerogel a, photographs b, c show the magnetic response of the CMC aerogel and carbon aerogel
The carbon aerogel was hydrophobic as shown in Fig. 6a; water drops could form on its surface and the WCA was 109°. In Fig. 6b, the CMC aerogel showed an apparent bright flame when placed in the flame of an alcohol burner. In contrast, carbon aerogel was non-flammable and showed no obvious flame when exposed to an alcohol burner as shown in Fig. 6c. This indicates that the carbon aerogel possessed flame resistance after pyrolysis treatment. This carbon aerogel has potential for use as a hydrophobic material and fire retardant. Further research on carbon aerogel oil adsorption, heat insulation and electric properties will be carried out in our future studies.

Photograph of hydrophobic carbon aerogel with water droplets on surface and inset showing water contact angle a. Photographs of CMC aerogel b and carbon aerogel c in alcohol burner flame
4 Conclusions
In summary, we prepared carbon aerogel by sol–gel, freeze-drying and pyrolysis. The as-prepared carbon aerogel possessed a channel-like three-dimensional network structure and showed a typical type-IV adsorption isotherm, which occurs because of the large amount of mesopores. Pyrolysis led to the decomposition of oxygen-containing functional groups, CMC crystalline structure damage and amorphous graphite generation, and provided the carbon aerogel with magnetic properties, which provides for potential application in recyclable adsorption materials, magnetic sensors and magnetic catalysts. The carbon aerogel was hydrophobic and flame retardant, which may allow for its application in hydrophobic materials and fireproofing.
References
R. Singh, R.K. Khardekar, D.K. Kohli, M.K. Singh, H. Srivastava, P.K. Gupta, Mater. Lett. 64, 843–845 (2010)
Y.J. Lee, J.C. Jung, J. Yi, S.-H. Baeck, J.R. Yoon, I.K. Song, Curr. Appl. Phys. 10, 682–686 (2010)
J. Wang, M. Chen, C. Wang, J. Wang, J. Zheng, Mater. Lett. 68, 446–449 (2012)
C. Wan, Y. Lu, Y. Jiao, C. Jin, Q. Sun, J. Li, Carbohydr. Polym. 118, 115–118 (2015)
Y.J. Lee, J.C. Jung, S. Park, J.G. Seo, S.-H. Baeck, J.R. Yoon, J. Yi, I.K. Song, Curr. Appl. Phys. 10, 947–951 (2010)
H.B. Chen, B.S. Chiou, Y.Z. Wang, D.A. Schiraldi, A.C.S. Appl, Mater. Interfaces 5, 1715–1721 (2013)
B. Chen, Y. Gao, Y. Bi, X. Luo, L. Zhang, Mater. Lett. 132, 75–77 (2014)
C. Wan, J. Li, Carbohydr. Polym. 134, 144–150 (2015)
Z.Y. Wu, C. Li, H.W. Liang, J.F. Chen, S.H. Yu, Angew. Chem. Int. Ed. 52, 2925–2929 (2013)
R. Lin, A. Li, L. Lu, Y. Cao, Carbohydr. Polym. 118, 126–132 (2015)
M. Xu, Y. Ao, S. Wang, J. Peng, J. Li, M. Zhai, Carbohydr. Polym. 128, 171–178 (2015)
H.L. Abd El-Mohdy, React. Funct. Polym. 67, 1094–1102 (2007)
S. Kakasi-Zsurka, A. Todea, A. But, C. Paul, C.G. Boeriu, C. Davidescu, L. Nagy, A. Kuki, S. Keki, F. Peter, J. Mol. Catal. B Enzym. 71, 22–28 (2011)
F. Zhang, Z. Pang, C. Dong, Z. Liu, Carbohydr. Polym. 132, 214–220 (2015)
J.F. Du, Y. Bai, W.Y. Chu, L.J. Qiao, J. Polym. Sci. Part B Polym. Phys. 48, 260–266 (2010)
J. Li, X. Wang, Q. Huang, S. Gamboa, P.J. Sebastian, J. Power Sources 158, 784–788 (2006)
X. Wang, X. Wang, L. Yi, L. Liu, Y. Dai, H. Wu, J. Power Sources 224, 317–323 (2013)
T.W. Lee, Y.G. Jeong, Carbohydr. Polym. 133, 456–463 (2015)
X. Wu, W. Jia, Chem. Eng. J. 245, 210–216 (2014)
G. Wang, H. Xu, L. Lu, H. Zhao, J. Energy Chem. 23, 809–815 (2014)
S. Lin, L. Chen, L. Huang, S. Cao, X. Luo, K. Liu, Ind. Crops Prod. 70, 395–403 (2015)
C. Vasile, G.G. Bumbu, R.P. Dumitriu, G. Staikos, Eur. Polym. J. 40, 1209–1215 (2004)
Q. Wu, W. Li, J. Tan, X. Nan, S. Liu, Appl. Surf. Sci. 332, 354–361 (2015)
Y. Zhang, C.P. Chai, Y.J. Luo, L. Wang, G.P. Li, Mater. Sci. Eng. B 188, 13–19 (2014)
Z. Liu, C. Lv, X. Tan, J. Phys. Chem. Solids 74, 1275–1280 (2013)
Acknowledgments
This work was supported by Heilongjiang Province outstanding youth science fund (JC201301).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yu, M., Li, J. & Wang, L. Preparation and characterization of magnetic carbon aerogel from pyrolysis of sodium carboxymethyl cellulose aerogel crosslinked by iron trichloride. J Porous Mater 23, 997–1003 (2016). https://doi.org/10.1007/s10934-016-0157-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-016-0157-4