
Abstract
SAPO-34 molecular sieves were synthesized hydrothermally using different combinations of amine agents [i.e., tetraethyl ammonium hydroxide (TEAOH) and diethylamine (DEA)]. XRD indicated the good crystallinity directed by single or mixed template for CHA structure. The morphology of crystals from SEM photograph was shown to be cubic shape of typical SAPO-34, but distinct in crystal size, increasing from 0.3–0.8 to 3.1–9.1 μm with the amount of DEA in the gel. NH3-TPD showed similar acid properties for SAPO-34 with TEAOH content from 1.0 to 0.1, but strong acidity synthesized with DEA only. 29Si MAS NMR demonstrated that Si species in framework with single DEA existed in large size of silicon islands, whereas dispersed finely in siliceous islands of smaller size with remarkable Si (nAl, 4-nSi, 0 < n < 4) environments in the samples using mixed template. In the methanol conversion to olefins over different SAPO-34 catalysts, the sample prepared with the mixtures of 50%TEAOH and 50%DEA showed the longest lifetime and high selectivity to C =2 ~C =3 .
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The introduction of silicon atoms in AlPO frameworks (SAPO) leads to the appearance of Brönsted acidity relevant for acid catalyzed reactions such as methanol-to-olefins (MTO), n-alkane cracking and hydrocracking, oxidative dehydrogenation of alkanes and so on. An increasing public attention has been focused on MTO process on account of the high crude oil price and the public awareness of the need for obtaining fuels from alternative sources (such as coal, nature gas and biomass) [1]. SAPO-34, with excellent performance in MTO conversion [2], is the best candidate for commercial applications.
The principle of the generation of protonic acidity of SAPOs, as known to all, may be explained by the introduction of Si in a theoretical framework of AlPO4. In fact, three potential mechanisms of Si substitution were proposed [3]. Si may replace Al (SM1), P (SM2), or Al–P pairs (SM3). As the formation of Si–O-P bonds is unlikely, such bonds formed in SM2 and 3 have to be modified by the replacement of P by Si. This gives rise to isolated silicon Si(4Al) and siliceous islands Si(nAl, 4-nSi, 0 < n < 4).
In SAPO synthesis, organic compound, i.e., template, plays the most important role. The action for an organic compound in AlPO or SAPO synthesis is well known for the role of structure directing agent (SDA), space filling and charge compensating. Its role was challenged, however, by the preparation of one specific structure with several SDAs, and conversely, by the existence of one SDA capable of producing more than one molecular sieves. The Si distribution depends on the number of framework charges, which is influenced by both the number of SDA molecules embedded within the cages and the Si atoms introduced within the framework [4]. Moreover, the elemental composition, local microscopic structure and morphology may also change with the use of different SDAs [5–7].
Such amines as Tetraethylammonium hydroxide (TEAOH) [8–10], diethylamine (DEA) [5], triethylamine (TEA) [11], morpholine (Mor) [4], or piperidine [12] have been used as SDA in SAPO-34 synthesis. Among them, TEAOH was widely used in lab for SAPO-34 synthesis with superior catalytic performance for MTO reaction. But its high price leads to the problem in practical use. However, DEA and Mor cost much lower than TEAOH, only 3.8 and 8.0% compared with that of TEAOH, respectively. In recent years, combinations of different SDA molecules has been used in the production of silicoaluminophosphates, such as TEAOH/morpholine for SAPO-34 [7], TPAOH/TMAOH for SAPO-37 and SAPO-40 [13], and DEA/di-iso-propylamine for SAPO-11 [14].
So far, research on the use of TEAOH/DEA as the SDA in SAPO-34 synthesis is relatively sparse. For each structure to be synthesized, there may exist an optimal templating agent or optimal combinations for a given mixed templating agent, which often remains to be found. The main purpose of the present work is to elucidate how the ratios of TEAOH/DEA influences the synthesis of SAPO-34 in the presence of TEAOH/DEA as the template and their catalytic performance in MTO conversion, and to obtain an excellent catalyst for MTO process.
2 Experimental
2.1 Catalyst preparation
Crystalline SAPO-34 was synthesized hydrothermally using different combinations of TEAOH (20 wt%) and DEA as the template. The reaction gel was made up from aluminum pseudoboehmite (70% Al2O3), ortho-phosphoric acid (85%), colloidal silica (30% SiO2), distilled water, TEAOH and DEA. The gel molar composition was Al2O3:P2O5:0.60SiO2:2xTEAOH:2(1 − x)DEA:53H2O (x = 1.0, 0.7, 0.5, 0.3, 0.1, 0). The prepared gel was transferred to a 1L titanium autoclave and heated to 100 °C for 24 h, and then 200 °C for 72 h. After the crystallization, the so produced solids were recovered by centrifugation, washed several times with distilled water and dried at 110 °C overnight. As-synthesized product was then calcined in air at 550 °C for 4 h in order to remove the organic templates in the pores of sample.
2.2 Characterization
The XRD patterns were recorded on a Rigaku D-max 2,200 X-ray diffractometer using Cu Ka radiations in the range of 2θ = 5°–60°. The BET surface areas were determined by nitrogen adsorption at 77.3 K on Micromeritics ASAP 2020M analyzer. The morphology was obtained using a JEOL JSM-6360LV scanning electron microscope. Elemental analysis for Al, P and Si of samples was done by sequential X-ray fluorescence spectrometer (XRF-1800, Schimadzu Corporation). NH3-TPD was performed on Micromeritics AutoChemII 2920. Typically, 200 mg of samples was pretreated at 550 °C for 2 h in He flow (40 mL/min) and was subsequently cooled to adsorption temperature of 100 °C. After contact of sample with gas mixture of NH3:He (10 vol.% of NH3) at the rate of 30 mL/min for 30 min, He flow was passed through the sample to remove weakly adsorbed NH3 molecules for 60 min. Finally, the He flow (40 mL/min) was passed through the sample with increasing temperatures up to 650 °C at the rate of 10 °C/min. 29Si MAS NMR spectroscopy measurement was conducted at resonance frequencies of 99.35 MHz, using a DRX500 spectrometer. The spinning rates of the samples at the magic angle were 4 kHz. The reference material for the chemical shift (in ppm) was tetramethylsilane (TMS). Differential thermal analysis (DTA) was performed on the calcined samples using TA instrument (SDTQ600) under an air flow (100 mL/min) at a heating rate of 10 °C/min from room temperature to 1,200 °C.
2.3 Catalytic reaction
The MTO conversion was carried out in a fixed bed at 425 °C under atmospheric pressure. 1.5 g samples (80–100 mesh), mixed with equi-mass of quartz, was packed on quartz wool plugging inside the stainless steel reactor (i.d.: 0.9 cm, length: 65 cm). Pure methanol was fed into preheater kept at 250 °C with a flow rate of 0.03 mL/min by an advection pump (2PB00C, Beijing Satellite Manufacturing Factory, China). The space velocity in WHSV was kept to 1 h−1. Nitrogen used as an inert diluent gas was co-fed with methanol into preheater at 60 mL/min. The analysis of the reaction products was performed on on-line gas chromatographs: Agilent GC 6890N (FID, Plot Al2O3/KCl) and GC 6820 (TCD, Plot Q).
3 Results and discussion
3.1 Structure, morphology and chemical composition
Hereafter, samples will be named making reference to the amount of TEAOH and DEA used in the synthesis; in this way, S-TEAOH, S-TEDE0.7, S-TEDE0.5, S-TEDE0.3, S-TEDE0.1, S-DEA will refer to SAPO-34 materials obtained with 2xTEAOH:2(1 − x)DEA (x = 1.0, 0.7, 0.5, 0.3, 0.1, 0) in gel, respectively (see in Table 1).
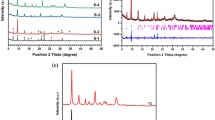
Figure 1 shows the X-ray diffraction patterns of synthesized SAPO samples prepared by different combinations of TEAOH and DEA. Accordingly, all the synthesized patterns exhibited typical repetitious powder diffraction patterns corresponding to CHA-structure of SAPO-34 reported in the literature [9]. It reveals that SAPO-34 was successfully crystallized not only with the pure SDAs, but also with their mixtures, being the crystallinity of the samples very similar to each other, with that for SAPO-34 obtained with DEA as the only SDA reaching the highest value.

XRD patterns of SAPO samples: a S-TEAOH, b S-TEDE0.7, c S-TEDE0.5, d S-TEDE0.3, e S-TEDE0.1, f S-DEA
Figure 2 displays the crystal morphology of samples observed by SEM photograph. As shown, the crystals of all samples are similar to be cubic shape of typical SAPO-34 [15] without obvious aggregations, but distinct in crystal size (see in Table 1). In the case of SAPO-34 synthesized with single TEAOH, homogeneous crystals with small particle size of 0.3–0.8 μm were obtained. Bycontrast, big crystals with different size, ranging broadly from 3.1 to 9.1 μm, were formed in the case of SAPO-34 synthesized with DEA only. This result means that the rate of crystal growth varied closely with the type of SDAs in the initial gel. Interestingly, as the molar content of DEA increased in the gel, a simultaneous increase of the crystal size was observed, indicating that these two opposite trends of TEAOH and DEA can be tailored to a certain point by using the mixtures of both SDAs. Thus, the nature of template used in synthesis not only influences the products structure patterns, but also determines the morphology of final crystals due to different rate of crystal growth. Moreover, the crystal particles have been observed smaller when using pure TEAOH of higher concentration (25 wt%) in our previous work. It is also worthwhile pointing out that the existence of large grains of S-DEA sample may be the reason for its sharp XRD-line in Fig. 1.

SEM images of SAPO-34 samples: a S-TEAOH, b S-TEDE0.7, c S-TEDE0.5, d S-TEDE0.3, e S-TEDE0.1, f S-DEA
The textural parameters (BET surface area and pore volume) through N2 adsorption isotherm are listed in Table 1. The surface area was maintained higher than 700 m2/g when TEAOH content (x) changed from 1.0 to 0.3. A further decrease of TEAOH content to 0.1 led to gradual decrease in surface area. When using DEA as SDA only, the sample S-DEA exhibited much lower surface area, as well as pore volume, in comparison with sample S-TEDE0.1 (TEAOH:DEA = 0.1:0.9). As regards the pore volume, S-TEDE0.5 possessed the highest value of 0.42 ml/g.
The corresponding final SAPO-34’s composition with respect to Si, Al and P based on (Si x Al y P z )O2 formula are obtained by XRF analysis also listed in Table 1. Compared to x = 0.130 for all starting gels, the value of the product samples was found to be higher with all mixed SDAs whereas lower with single TEAOH or DEA, which means more amount of Si was incorporated into the framework of SAPO molecular sieves and/or remained as amorphous silica phase on extra-framework. Considering the good crystallinity from XRD data in Fig. 1, it can be deduced that silica existing as amorphous phase on extra-framework is unreasonable. This result is not consistent with that for Mor/TEAOH synthesis of SAPO-34 [7], which was reported that parts of silica remained in extra-framework as amorphous phase confirmed by the decrease intensities in XRD and NH3-TPD curves.
3.2 Acidity (NH3-TPD and 29Si MAS NMR)
The acid sites concentration and strength of the calcined samples were characterized by temperature programmed desorption of ammonia (NH3-TPD) technique (Fig. 3). Two TPD peaks for samples at 200–250 and 450–500 °C are observed in Fig. 3a–e. The desorption temperature signifies the strength of acid sites, given that the stronger acid sites requires a higher desorption temperature, and the area under the NH3 desorption curve indicates the amount of ammonia desorbed, which is proportional to the acidity [14]. The first peak, corresponding to weak acid sites, is attributed to the P-OH hydroxyl groups not fully linked to AlO4 tetrahedra [16]. It is said to be useless for methanol conversion to light olefins, but there reported no charts to illustrate it. Thus, the NH3-TPD test was also performed on deactivated samples after reaction in comparison with that before reaction (Fig. 4). It was obvious that the second peak of deactivated sample was absolutely missing and the first peak maintained as that before reaction. Therefore, the second desorption peak should be active for MTO conversion, which is assigned to the bridging hydroxyl groups, i.e., -SiOHAl-, formed by replacement of phosphorous by silicon. The disappeared second peak for deactivated samples can be explained that the strong acid sites was gradually covered or hindered by coke formation during the reaction procedures, simultaneously with obvious decrease in surface area.

NH3-TPD Profiles of SAPO-34 samples: a S-TEAOH, b S-TEDE0.7, c S-TEDE0.5, d S-TEDE0.3, e S-TEDE0.1, f S-DEA

NH3-TPD Profiles of deactivated S-TEDE0.5
According to the peaks in Fig. 3, the weak and strong acidity of samples are derived and shown in Table 1. It shows similar acid properties between these samples with TEAOH content from 1.0 to 0.1 both in acid sites concentration and strength. However, for sample S-DEA with single DEA, the first peak at low temperature seems difficult to be identified and the second peak shifts to high temperature at ca. 650 °C, simultaneously with a decreased area, which can be explained by the existence of large size of silicon islands in the framework from 29Si MAS NMR spectra given later in Fig. 5.

29Si MAS NMR spectra of SAPO-34 samples: 1 Si (4Al), 2 Si (3Al), 3 Si (2Al), 4 Si (1Al), 5 Si (0Al)
Figure 5 shows the silicon distribution from 29Si MAS NMR spectra and the deconvolution results by Gaussian Fit are concluded in Table 2. As shown, the spectra could be deconvoluted into five peaks centered at −90, −94, −100, −105 and −112 ppm due to Si(4Al), Si(3Al, 1Si), Si(2Al, 2Si), Si(1Al, 3Si) and Si(4Si) environments, respectively. As to sample S-DEA, correlating with its low Si content of 0.110, the existence of large size of silicon islands and serious Si–Al domains are the reasons for its strong acid strength and low concentration. It can be confirmed from its dominant silicon resonances due to Si (4Si), associated with less intense signals due to Si(3Al, 1Si), Si(2Al, 2Si), Si(1Al, 3Si), and the broadness of the peak at −90.0 ppm [17]. As to S-TEAOH, it possesses high percent of peak resonance due to Si (4Al). Therefore, although with low content of Si in framework, sample S-TEAOH displayed good acid properties. Different from the two samples with single SDAs, samples S-TEDE series show remarkable Si(nAl, 4-nSi, 0 < n < 4) environments also with signals due to Si (4Al) and Si (4Si) presenting. As is known, the smaller size of Si island indicates more number of Si island, which generates more Si (nAl, 4-nSi, 0 < n < 4) sites at the border of Si domain. Hereby, although incorporating higher content of silicon into framework when using mixed TEDE, the Si species in S-TEDE series dispersed finely in siliceous islands of smaller size instead of large size of silicon islands and/or amorphous Si species. As a result, S-TEDE series maintained sufficient number of acid sites as shown in Table 2.
3.3 Thermal stability
The thermal stability of samples is estimated by DTA experiments (Fig. 6). All the synthesized samples have an endothermic peak in the DTA curves at ca. 100 °C, which is attributed to the physically adsorbed water associated with samples. Another exothermic peak is also observed at the high temperature nearly up to 1,100 °C, which is attributable to the skeleton collapse of samples, indicating that all the synthesized samples exhibited good thermal stability.

DTA curves of calcined SAPO-34 samples
3.4 MTO performance
The calcined samples were tested as catalyst in MTO conversion at 425 °C. Figure 7 shows the yield of products with time on stream (TOS) on S-TEDE0.5. The yield of ethylene increased gradually with TOS, getting maximum value of about 56.08%, and decreased afterward. The yield of propylene and butane generally shows slight decrease during the reaction. It is worth to point out that saturated hydrocarbons propane was prominent in products in the initial stage of reaction and dropped rapidly with TOS to less than 3%. This phenomenon can be explained that at the activation period, the formation of active intermediates such as hexamethylbene (hexaMB) released some vacant H+ which favors the hydrogen transfer reaction [18]. Thus, the yield to light olefins (C =2 -C =4 ) was observed a little low in initial stage, and later maintained over 90% until catalytic activity abruptly decreased by deactivation of catalyst due to coke formation. It was noted above that weak acid sites was useless for olefins formation, but the conversion of methanol to DME can occurred on weak acid sites. Therefore, after deactivation when most strong acid sites were covered or hindered by coke formation, the yield of light olefins decreased rapidly, simultaneously with steep increase of DME.

Distribution of the products with TOS on S-TEDE0.5 sample
The catalytic performances on all synthesized samples are shown in Table 3 and Fig. 8. As shown, all these samples except for S-DEA had complete conversion of methanol and mostly conversion to light olefins (>90%). S-DEA catalyst, synthesized with single DEA, not only has incomplete methanol conversion and low light olefins, but also shows the shortest catalyst lifetime (herein means the time sustaining catalyst activity until intermediate product DME exists in products). These results are mainly due to its large crystal size and strong but low number of acid sites [19, 20]. Too strong acid strength, associated with large crystal size, can promote hydrogen transfer reaction to form aromatics and branched hydrocarbons, which could be trapped inside the cavities as coke and result in fast coke formation. Thus, S-DEA catalyst kept active only for 115 min and high amount of propane (7.98%) was observed in product.

Time course of the DME selectivity on SAPO-34 catalysts
As to samples S-TEAOH and S-TEDE series with similar high selectivity to light olefins, the most striking feature is the differences in the catalyst lifetime. Sample S-TEDE0.5 maintained activity without intermediate DME in products for 545 min, after which DME emerged out and increased rapidly. The lifetime was in the order of S-TEDE0.5 > S-TEDE0.3 > S-TEDE0.7 > S-TEAOH > S-TEDE0.1. In association with the previous analysis results, there is no obvious discrepancy in acidity (Fig. 3), but shows significant differences in crystal size between these catalysts. Thus, it can be deduced that the differences in lifetime depend mostly on the crystal size. The crystal size decreased in the order of S-TEDE0.1 > S-TEDE0.3 > S-TEDE0.5 > S-TEDE0.7 > S-TEAOH. It appears from the results that neither small nor large crystal size is favorable for catalysts to prolong the lifetime. In the case of S-TEAOH samples, they had too short diffusion length for reaction intermediate to react to olefins. Whereas, as to S-TEDE0.1, the crystal size was proved to be lager for MTO conversion. Therefore, the longest lifetime of S-TEDE0.5 was due to its optimal average crystal size of about 1.15 μm.
4 Conclusion
SAPO-34 crystals were well-crystallized by hydrothermal method using different combinations of TEAOH and DEA (named TEDE) as template. The morphology of samples, was similar to be cubic shape of typical SAPO-34, but distinct in crystal size, increasing with the content of DEA in the gel. It indicates that the nature of template used in synthesis determines the morphology of final crystals due to different rate of crystal growth. From NH3-TPD, similar acid properties was observed for SAPO-34 with TEAOH content from 1.0 to 0.1, but strong acidity synthesized with DEA only, which was due to existence of large size of silicon islands from 29Si MAS NMR. In MTO conversion, all the SAPO-34 catalysts showed activity. The catalyst obtained using the mixture of 50% TEAOH and 50% DEA gave the longest lifetime due to optimal crystal size.
References
F.C. Patcas, J. Catal. 231, 194 (2005)
M. Stöcker, Micropor. Mesopor. Mat. 29, 3 (1999)
G. Sastre, D.W. Lewis, C.R.A. Catlow, J. Phys. Chem. B 101, 5249 (1997)
R. Vomscheld, M. Briend, M.J. Peltre, P.P. Man, D. Barthomeuf, J. Phy. Chem. 98, 9614 (1994)
G.Y. Liu, P. Tian, J.Z. Li, D.Z. Zhang, F. Zhou, Z.M. Liu, Micropor. Mesopor. Mat. 111, 143 (2008)
A.M. Prakash, S. Unnikrishnan, J. Chem. Soc. Faraday Trans. 90, 2291 (1994)
Y.J. Lee, S.C. Baek, K.W. Jun, Appl. Catal. A Gen. 329, 130 (2007)
S. Kvisle, R. Wendelbo, H. Ren, US Pat. 5,663, 471 (1994)
M. Popova, C. Minchev, V. Kanazirev, Appl. Catal A Gen. 169, 227 (1998)
M. Mertens, K.G. Strohmaier, European Pat. EP1451105 (2004)
Z.Z. Liu, G.Y. Cai, C.Q. Chang, L.X. Yang, Z.Z. Wang, J.S. Luo, Y.J. Chang, R.M. Shi, Z.Q. Jiang, C.L. Sun, CN Pat. 1087292 (1994)
E. Dumitriu, A. Azzouz, V. Hulea, D. Lutic, H. Kessler, Micropor. Mat. 10, 1 (1997)
J.P. Lourenco, M.F. Ribeiro, F.R. Ribeiro, J. Rocha, Z. Gabelica, Stud. Surf. Sci. Catal. 84, 867 (1994)
P. Liu, T. Ren, Y.H. Sun, Micropor. Mesopor. Mat. 114, 365 (2008)
J.W. Park, J.Y. Lee, K.S. Kim, S.B. Hong, G. Seo, Appl. Catal A Gen. 1, 36 (2008)
B. Parlitz, E. Schreier, H.L. Zubowa, R. Eckelt, E. Lieschke, R. Fricke, J.Catal. 155, 1 (1995)
M. Briend, R. Vomscheid, M.J. Peltre, P.P. Man, D. Barthomeuf, J. Phy. Chem. 99, 8270 (1995)
U. Olsbye, M. BjØrgen, S. Svelle, K.P. Lillerud, S. Kolboe, Catal. Today 106, 108 (2005)
L. Travalloni, A.C.L. Gomes, A.B. Gaspar, MAP da Silva. Catal. Today 133–135, 406 (2008)
D. Chen, K. Moljord, T. Fuglerud, A. Holmen, Micropor. Mesopor. Mat. 29, 191 (1999)
Acknowledgments
The authors gratefully acknowledge the financial support of the National Key Technology Program of China (No. 2006BAE02 B02) and Yankuang Energy R&D Co., Ltd., Shanghai.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ye, L., Cao, F., Ying, W. et al. Effect of different TEAOH/DEA combinations on SAPO-34’s synthesis and catalytic performance. J Porous Mater 18, 225–232 (2011). https://doi.org/10.1007/s10934-010-9374-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10934-010-9374-4