
Abstract
The forkhead box (FOX) proteins are a family of transcription factors that interact with DNA via a winged helix motif that forms part of the forkhead domain. The FOXP (FOXP1–4) subfamily is unique in the family in that the forkhead domains of these proteins are able to dimerise via domain swapping. In this event, structural elements are exchanged via extension of the hinge loop region. Despite the high sequence homology among the FOXP subfamily members, the stability of their forkhead domain dimers varies, with FOXP3 forming the most stable dimer. An amino acid difference is observed in the hinge region of the FOXP subfamily where a tyrosine in all members is replaced with a phenylalanine in FOXP3. In this work, the role of phenylalanine at this position in the hinge region was investigated. This was done by creating the Y540F variant of the FOXP2 forkhead domain. The effect of the Y540F mutation on the structure, dimerisation propensity and DNA binding ability of the FOXP subfamily was investigated. The mutation altered the structure of the protein by decreasing the disorder of the backbone as measured by circular dichroism spectroscopy and by altering the local environment of the hinge region as measured by tryptophan fluorescence. The propensity of the forkhead domain to form a dimer was improved ~9.5 fold by the mutation. This was attributed to increased hydrophobicity at the dimer interface as well as altered tension in the hinge loop region. DNA binding assays indicated that the affinity for DNA was decreased by the mutation. Taken together, these findings suggest that domain swapping may modulate DNA binding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The forkhead box (FOX) superfamily of transcription factors share homology only in their highly conserved DNA binding, winged-helix forkhead domains (FHDs) [1, 2]. The FOX proteins have largely diverse yet important functions including roles in the immune system [3, 4], metabolism [5], regulation of cellular proliferation and differentiation [1, 6], ageing [6] and embryonic development [7, 8]. Mutations in the forkhead domain have been linked to a wide variety of human diseases for example alopecia (FOXN1) [9], thyroid agenesis (FOXE1) [10], Axenfeld-Rieger syndrome (FOXC1) [11, 12], tumorigenesis and intellectual disability (FOXP1) [11–13], a speech deficit disorder (FOXP2) [14] and IPEX (immune dysregulation, polyendocrinopathy, enteropathy and X-linked) syndrome (FOXP3) [15]. Since these mutations are located in the DNA binding domain, the FHD, they compromise the DNA binding capacity of FOX transcription factors. The FHD also contains a nuclear localisation signal and thus, mutations in this region may affect patterns of subcellular localisation [15–18]. For example, the FOXP2 speech-related mutation (R553H) shows complete abolishment of DNA binding ability and altered nuclear localisation [14]. One FOXP3 IPEX mutation (A384T) affects DNA binding, whereas the F371C and F373A IPEX mutations are shown to affect domain swapping and suppressor function [15, 16]. To date, no known FOXP4 mutations have been linked to disease.
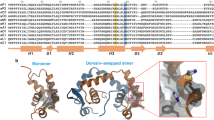
Structurally, the isolated FHDs of the FOX proteins are monomeric whether in the presence or absence of DNA [19] and this is reflected in the solved crystal or NMR structures for example, FOXA3 [20], FOXC2 [21], FOXD3 [22], FOXK1a [23], FOXO3a [24], FOXO4 [25], FOXM1 [26], FOXQ1 [27] and FOXO1 [28]. The only exception identified to date is the P-subfamily where the crystal structures of the isolated forkhead domains of FOXP2 and FOXP3 show the formation of domain-swapped dimers [16, 29].
Domain swapping is a type of oligomerisation event in which specific regions of proteins are exchanged to form an intertwined dimer [30]. The physiological relevance of domain swapping is still unclear and some domain-swapped proteins may merely be artefacts of the high concentrations needed for crystallisation [31, 32]. The event has been associated with aggregation [33]. Furthermore it has been implicated in physiological processes including neurodegenerative diseases [34–36], the evolution of oligomeric proteins [37] as well as the regulation of protein activity [33, 38–40]. Protein activity can be regulated through changes in the concentration of the protein which will regulate what proportion of monomer or domain swapped dimer is present. In this regard, macromolecular crowding in the cell, for example, could increase the local concentration of a particular protein [41–43], thereby increasing its propensity to domain swap and carry out its specific function. Other factors such as the length [44–46], flexibility [46–48] and hydrophobicity [45, 49, 50] of the hinge-loop region have been shown to affect domain swapping propensity.
In the FOXP2 and FOXP3 domain swapped dimers, helix H3 and β-strands S2 and S3 are exchanged [16, 29, 51]. The hinge-loop region (H4) changes conformation upon domain swapping to form an extended helix H2 in the domain swapped dimer [16, 29]. Ala539 in FOXP2 (and its equivalent alanine in other FOXP proteins) is located in the hinge-loop region (Fig. 1). In most other FOX proteins, this alanine is substituted with a proline. Mutation of Ala539 to proline in FOXP2 results in the FHD existing exclusively in the monomeric form [29]. It is thought that the rigid proline residue prevents extension of the hinge-loop region (H4) which is required for domain swapped dimer formation. The consequence of this is that only the FHDs of the FOXP family members are able to dimerise.

Sequence alignment of the FOXP family. Secondary structural elements are indicated above the sequence. FOXP2 numbering is shown above the sequence and FOXP3 numbering is shown below. ClustalX version 2.0 [57] was used to perform the alignment and the default colouring scheme was used. The hydrophobic core residues that stabilise the dimer interface are highlighted (black circles above sequence) with the hinge-loop region indicated in the boxed region. The tyrosine (Y540) residue in the hinge-loop region which is the focus of this study is indicated by the arrow below the sequence. The alanine (A539) residue found to be crucial to domain swapping in the FOXP subfamily is indicated by the black box above the sequence
Although the highly conserved FHDs of FOXP1, FOXP2 and FOXP3 have all been shown to dimerise [16, 29, 51], the FOXP3 dimer is considerably more stable than the FOXP2 [29] or FOXP1 [51] dimer and furthermore the FOXP3 dimeric form is reported to be necessary for its suppressor function [16]. Thus a comparison between the FOXP2 and FOXP3 dimers might give clues as to what improves the stability of the FOXP3 dimeric form compared to FOXP2. This will have implications for the functionality of the proteins. A network of hydrophobic core residues is suggested to control the formation of the FOXP3 domain swapped dimer: Tyr364, Trp366, Phe367, Phe373, Phe374 and Trp381 (Bandukwala et al. [16]). There is a similar corresponding network in FOXP2 (Fig. 2) (Stroud et al. [31]). Sequence differences in this hydrophobic network and/or in the hinge loop region may account for the difference in stabilities of the two domain swapped dimers.

The FOXP2 domain swapped dimer interface. Residues Tyr531, Trp533, Phe534, Tyr540, Phe541 and Trp548 are found at the dimer interface and form part of the hydrophobic network which is thought to be involved in stabilising the domain swapped dimer [25]. Tyr540 is highlighted. Images were generated using PyMOL. PDB code 2A07
Comparison of the sequences of the hinge loop region of the FOXP subfamily members (Fig. 1) shows only two amino acid differences. These are located at positions Phe373 and Asn376 in FOXP3 and are conserved as tyrosine and arginine respectively in all the other members of the FOXP subfamily. Phe373 not only forms part of the hinge loop region but is also buried deep in a stabilising hydrophobic pocket at the dimer interface, implicating this residue in dimer stability. A F373A mutation in the FOXP3 FHD has been linked to IPEX syndrome [15]. This IPEX-causing mutation causes a shift in the monomer–dimer equilibrium towards the monomer by disrupting the hydrophobic core of the domain swapped dimer [16]. The work presented here investigates the significance of having a phenylalanine at this position for dimerisation and DNA binding of the FOXP forkhead domain, particularly because of the reported difference in stability of the dimer across the FOXP family. In FOXP2 there is a tyrosine residue (Tyr540) (Fig. 2) located at the equivalent position to Phe373 in FOXP3. Therefore, in this study, the Y540F FOXP2 FHD variant was created in order to establish the role of having a phenylalanine located both in the hinge loop and at the dimer interface in the FOXP subfamily and to establish whether this phenylalanine is responsible for the greater propensity that FOXP3 has for dimerisation compared to FOXP2.
2 Materials and Methods
2.1 Site-Directed Mutagenesis, Protein Expression and Purification
The pET-30 LIC plasmid encoding the FOXP2 FHD hexahistidine-tagged fusion protein was used as a template for site-directed mutagenesis. The primers used to generate the mutant were designed using Primer X (http://www.bioinformatics.org/primerx/) and had the following sequence: 5′ CACGGACATTTGCGTTCTTCAGGCGTAATGCAGCAAC3′. The mutation codon is in bold. The mutant primer and its reverse complement were synthesised by Inqaba Biotec (Pretoria, South Africa). The Y540F FOXP2 FHD mutant was generated using the Quikchange® Lightening Kit (Stratagene, La Jolla, CA). The plasmid was sequenced by Inqaba Biotec to ensure that the mutation was incorporated and that no undesirable mutations were introduced. The wild type and mutant proteins were expressed in T7 Express Competent Escherichia coli cells (New England Biolabs, Ontario, Canada). The proteins were purified on nickel charged IMAC resin (GE Healthcare) and stored in 20 mM Tris–Cl pH 7.6, 1 mM DTT and 150 mM NaCl. Contamination by E. coli DNA non specifically binding to the transcription factor was eliminated using a high salt wash (1 M NaCl) on the nickel column. This reduced the A260:A280 to acceptable levels indicating negligable DNA contamination. Purity was assessed using SDS-PAGE stained with Coomassie Brilliant Blue R-250.
2.2 Spectroscopy
Far-UV circular dichroism (CD) spectra were collected with a Jasco J-810 spectropolarimeter at 20 °C. CD spectra were collected in triplicate, corrected for solvent, and converted to mean residue ellipticity [θ] (degrees square centimetres per decimole per residue); [θ] = (100θ)/(Cnl), where θ is the measured ellipticity (millidegrees), C is the protein concentration (millimolar), n is the number of residues and l is the path length in centimetres. The spectra were compared using the Dichroweb server [52].
Fluorescence measurements were collected on a Perkin-Elmer LS50B luminescence spectrofluorimeter at 20 °C. The emission spectra were recorded in triplicate from 300 to 450 nm using an excitation wavelength of 295 nm which selectively excites the three tryptophan residues (Trp533, Trp548 and Trp572).
2.3 Size-Exclusion Chromatography
A Hiload™ 16/600 75 pg size-exclusion column (GE Healthcare) was used to determine the relative amounts of monomer and dimer in both the wild-type FOXP2 FHD and the Y540F variant. Concentrations ranging from 5 to 300 μM of both wild-type FOXP2 FHD and the Y540F variant were left to equilibrate for 24 h at 4 °C prior to loading onto the column. The protein was detected by absorbance at 280 nm. The dissociation constant of dimerisation was calculated using the formula: [monomer]2/[dimer]. The area under the peaks of the monomer and dimer elution curves were used to calculate the relative amounts of monomer and dimer. The data was fitted with the linear regression method in Sigmaplot.
2.4 DNA Binding
The double-stranded FOX cognate sequence, 5′-AACTATGAAACAAATTTTCCT-′, synthesised by Integrated DNA Technologies (Whitehead Scientific, South Africa) was used in all DNA binding studies as it has been shown to bind to the FOXP2 FHD [29] as well as to other FOX proteins [53]. The core binding sequence is underlined. This specific flanking sequence was chosen as it has been shown to bind to the FOXP2 FHD in the crystal structure [29].
2.4.1 Electrophoretic Mobility Shift Assay
The FOXP2 FHD proteins were added to DNA at protein:DNA ratios ranging between 1:2 and 3:1, in binding buffer: 20 mM Tris–Cl, pH 7.6, 100 mM NaCl, 1 mM DTT, 1 mM MgCl2 and 20 % glycerol. The protein and DNA were incubated on ice for 30 min to allow for binding. The protein-DNA complexes were resolved on a 10 % acrylamide gel at 4 °C at 100 V for 1.5 h. The gel was visualised under ultraviolet light after being stained with 0.5 μg/ml ethidium bromide. The gels were analysed by densitometry using the LabWorks Image Acquisition and Analysis Software version 4.6 (UVP Bio-Imaging) to quantify the proportion of DNA bound to the FOXP2 FHD proteins. Quantitation was performed in triplicate on non-saturated images. The proportion of bound DNA was calculated as follows: % bound DNA = (100 %) − (% unbound DNA); where the sum of the intensity of all bands in a single lane is equal to 100 %. The data were collected in triplicate and fit to a one site saturation ligand binding model: \(y = \frac{{B_{\hbox{max} } x}}{{K_{d} + x}}\) in order to obtain the K d of DNA binding.
3 Results
The location of Tyr540 in the FOXP2 FHD dimer is of significance for two reasons: (1) it is located in the hinge loop region of the domain swapped dimer and (2) it is located at the hydrophobic dimer interface. In this study, we investigated structural the role of Tyr540 in the FOXP2 FHD and whether it influenced DNA binding. We created the Y540F FOXP2 FHD variant that resembles FOXP3 at this position and compared it structurally to the wild type by investigating secondary and tertiary structure as well as propensity to form a dimer. We also examined changes to structure upon DNA binding between the wild type and the variant and we compared the DNA binding affinity of the two.
3.1 Structural Integrity of the FOXP2 FHD
The secondary and tertiary structure of the wild type and Y540F FOXP2 FHD were compared (Fig. 3) to determine if the mutation had resulted in any structural changes to the protein. The far-UV CD spectra of the Y540F variant and the wild type are not superimposable. Most of the difference between the two spectra can be seen in the 190–215 nm range which represents the π → π* transition of the amide bond [54]. Further analysis of the spectra using Dichroweb [52] shows that while incorporation of the mutation does not appear to change the helical content of the protein, the variant does show an increase in β-sheet content by nearly 20 % compared to the wild type with a corresponding decrease in random coiled structure (Fig. 3b). The replacement of Tyr540 with a phenylalanine therefore appears to influence the backbone structure of the FHD by making it more structured overall.

a Far-UV circular dichroism spectra of 8 μM WT FOXP2 FHD (solid line) and the Y540F variant (dashed line) in 20 mM Tris, 150 mM NaF, 1 mM DTT, pH 7.6. The spectra were converted from millidegrees to mean residue ellipticity ([θ]), corrected for buffer and smoothed. b Secondary structural content analysis of the FOXP2 FHD. The data for the wild type and Y540F variant were obtained from the Dichroweb analysis of the CD spectra. NRMSD = 0.07 for WT and NRMSD = 0.150 for the Y540F mutant. The data for the FOXP2 monomer and dimer crystal structure were obtained from the PDB file 2A07 [29] and the data for the FOXP3 dimer crystal structure was obtained from PDB file 3QRF [16]. Image generated using Sigmaplot. c Fluorescence emission spectra of 4 μM WT FOXP2 FHD (solid line) and the Y540F variant (dashed line) in 20 mM Tris–Cl pH 7.6, 150 mM NaF, 1 mM DTT when excited at 295 nm. Images were generated using Sigmaplot
The fluorescence spectra indicate that the tryptophan fluorescence of the variant is enhanced about 1.5 times compared to the wild type. There are no wavelength shifts in the spectra however, as they both peak at 335 nm. This implies that although the polarity of the environment of the tryptophan residues is not altered by the mutation, there is a change in the local packing of the tryptophan residues when Tyr540 is replaced by a phenylalanine.
3.2 Dimerisation Propensity of the FOXP2 FHD
Size exclusion chromatography was performed on increasing concentrations of both wild type and Y540F variant (Fig. 4). The peaks that were eluted from the size exclusion column corresponded to the size of either monomeric or dimeric FOXP2 FHD respectively. The size exclusion profiles show that while the wild type FOXP2 FHD remains almost exclusively monomeric at concentrations up to 100 µM, the variant shows an increased propensity to dimerise. This can be seen in the estimated K d for dimerisation which is ~246 µM for the variant and ~2.4 mM for the wild type. Therefore the single replacement of Tyr540 with a phenylalanine causes an over 9.5 fold increase in the propensity of the FOXP2 FHD to dimerise. The purified monomer and dimer fractions of the Y540F FOXP2 FHD show identical fluorescence spectra (Fig. 3c), indicating that there is no tertiary structural change to the protein upon dimerisation.

Size-exclusion elution profiles of the a WT FOXP2 FHD and b Y540F variant. Increasing concentrations of protein were subjected to size-exclusion chromatography using a 16/600 Hiload Superdex 75 prep grade column (GE Healthcare) equilibrated with 20 mM Tris–Cl pH 7.6, 150 mM NaCl, and 1 mM DTT at 20 °C. c Tryptophan fluorescence spectra of the separated monomer and dimer fractions of Y540F FOXP2 FHD. d The dimer dissociation constant was calculated to be 246 μM for the FOXP2 Y540F mutant (closed circles) and 2389 μM for the WT FOXP2 FHD (open circles) using the formula: K d = [monomer]2/[dimer]. The data were fit using the linear regression method in Sigmaplot. R2 = 0.994 for the data fit for the Y450F mutant while R2 = 0.958 for the data fit for the WT FOXP2 FHD. Images were generated using Sigmaplot
3.3 DNA Binding
Both the wild type FOXP2 FHD and the Y540F variant were incubated at increasing concentrations in the presence of DNA for 30 min at 4 °C following which, electrophoretic mobility shift assays (EMSAs) were performed on the mixtures. A representative EMSA gel is shown in Fig. 5a. The free, unbound DNA migrates the furthest in the gel as seen in the control (lane 1). When protein is added, the amount of free DNA decreases and either one or two concentrated bands appear higher up in the gel. These bands are likely indicative of monomer or dimer associated with the DNA which accounts for the retarded movement of these complexes through the gel. The bands are not very sharp, however and the smearing can be attributed to the weak interaction that the FHD has with DNA in the absence of the remainder of the protein [19]. The protein sample of both the wild type and Y540F variant of FOXP2 FHD that was used for the EMSAs was pure as seen in the SDS PAGE lanes in Fig. 5b. This confirms that the smearing and banding evident in the EMSA is as a result of the various species of FOXP2 FHD–DNA complex present in the solution and not as a result of other protein contaminants associating with the DNA.

a EMSA on a 15 % native polyacrylamide gel. All lanes contain cognate DNA at 10 μM. Lane 1 contains free cognate DNA; lanes 2, 4, 6 and 8 contain 10, 20 and 30 μM of WT FOXP2 FHD, respectively. Lanes 3, 5, 7 and 9 contain 10, 20 and 30 μM of the Y540F FOXP2 FHD mutant, respectively. b 15 % SDS-PAGE gel indicating that both the wild type and Y540F samples were pure. c Densitometry analysis. The percentage of bound DNA was calculated using: fraction of bound DNA = 1 − fraction of unbound DNA × 100 where fraction unbound DNA is given by the densitometry of the bottom band in the EMSA gel. The fit of wild type data (solid line) to a single site binding model had an R2 of 0.95 and gave a K d of 21.0 ± 7.1 µM while the data for the Y540F mutant (dashed line) fit with an R2 of 0.85 and gave a K d of 32.1 ± 11.1 µM. Images were generated using Sigmaplot
Inspection of the representative gel in Fig. 5a indicates that there is a greater proportion of dimeric form interacting with the DNA in the variant compared to the wild type. This confirms the results from Fig. 4 that show that the Y540F variant is more prone to dimerise than the wild type. Despite this, the amount of total DNA interacting with the protein appears to be greater with the wild-type than the variant. In order to make a qualitative comparison of the binding between the wild type and the variant, a DNA binding curve was constructed (Fig. 5c) and fit to a single site binding model. Due to the smearing in the lanes, it is not possible to accurately quantify the amount of DNA bound specifically to the monomer or the dimer using densitometry. Rather, the amount of unbound DNA was quantified relative to the DNA-only control and from this, the percentage of total bound DNA (be it to the monomer or the dimer or at some intermediate condition) could be inferred. The fit of the data to a single site binding model had relatively low R2 values indicating that from this data, the protein concentration does not accurately demonstrate the amount of variation in the bound DNA. The degree of error is consistent throughout the EMSA replicates and could be attributed to inaccuracies in the densitometry or staining procedure used. Thus the K d values of 21.0 µM for the wild type and 32.1 µM for the variant are unlikely to be absolute values and should be referred to as apparent values. Nevertheless, from a qualitative perspective, it does appear that the wild type has a greater affinity for the DNA than the variant and this is supported by the apparent K d values.
4 Discussion
The FOXP proteins are the only FOX proteins whose FHDs have been shown to form a domain-swapped dimer. This is confirmed by the crystal structures of FOXP2 [29] and FOXP3 [16]. Furthermore, although the presence of a FOXP1 domain swapped dimer has not been confirmed, the FOXP1 FHD has also been shown to exist as a mixture of monomer and dimer in solution [51] and it is likely that this dimer is also domain swapped. The exclusive ability of these FOXP proteins to form a domain swapped dimer within the FOX family is due to a unique feature of their sequence where a conserved proline is replaced with an alanine allowing the extension of helix H2 via a hinge region. It is remarkable to note, however, that even within the FOXP subfamily, the propensity to form the dimer varies, with FOXP3 being more prone to dimerise than the other FOXP members [16] despite high sequence conservation between all members. It is in the interest of this work, therefore, to investigate this difference.
Hinge regions are crucial to domain swapping and changes in the length and flexibility of these regions have been shown to either support or disrupt domain swapping [46, 55]. The FOXP hinge region consists of the residues FAYFRR in all family members except for FOXP3 where the sequence is FAFFRN (Fig. 1). These two amino acid differences in the hinge region (Phe373 and Asn376 in FOXP3) may be responsible for the greater stability of the FOXP3 dimer. Because the point mutation of Phe373 to alanine has been implicated in IPEX syndrome and furthermore it has been shown to reduce dimerisation propensity [16], the corresponding tyrosine in FOXP2 (Tyr540) was replaced with a phenylalanine in this study and the effect on dimerisation and DNA binding was observed.
The Y540F mutation appears to have an effect on the structure of the FOXP2 FHD (Fig. 3). The backbone appears to become more structured upon mutation, causing an increase in β structure compared to the wild type. Since the helical percentage remains unchanged, this change in secondary structure simply reflects an increase in backbone ordering in the variant. Comparison of the crystal structures of the FOXP2 [29] and FOXP3 [16] domain swapped dimers supports this observation. The FOXP2 dimer structure, which has a tyrosine at position 540, has lower β strand content and higher random coil content than the FOXP3 dimer structure which has a phenylalanine at the equivalent position (Fig. 3). Thus a phenylalanine at position 540 results in ordering of the backbone of the FOXP2 FHD compared to when tyrosine is at that position. Because the concentration of protein used for the CD spectra was low enough that no significant proportion of dimer was present in either the wild type or the variant as determined by size exclusion chromatography (Fig. 4), it can be concluded that the observed structural changes are also occurring in the monomeric state. The most noteworthy difference between the secondary structural analysis of the monomeric FOXP2 FHD crystal structure and the Dichroweb analysis of the CD spectra presented in Fig. 3, is that the helical content of the crystal structure is approximately double that obtained from either the wild type or the variant in solution. This could possibly be attributed to the fact that the crystal structure was solved in the presence of DNA and perhaps DNA binding increases the helicity of the structure which leads to an increase in backbone order. In fact, a trend can be seen in Fig. 3b where the higher the dimer propensity, the greater the ordering of the backbone and this is more pronounced when in the presence of DNA. The wild type monomer in the absence of DNA shows the most disorder followed by the Y540F variant that shows increased dimer propensity, followed by the monomer in the presence of DNA, followed by the wild type dimer in the presence of DNA, followed by the FOXP3 dimer in the presence of DNA which has the highest propensity to dimerise.
The structural difference between the wild type and the Y540F variant is not only evidenced by the change in backbone structure. It can also be seen by the change in tryptophan fluorescence which implies a change to the local environment of the tryptophan residues upon the mutation (Fig. 3c). As with the CD spectra, the fluorescence spectra were measured at a concentration at which both wild type and variant would be almost entirely monomeric. This means that the changes in tertiary structure evident upon mutation are occurring in the monomeric form. The replacement of Tyr540 with a phenylalanine causes fluorescence enhancement due to a change in the local environment of the tryptophan residues. Quenching of tryptophan fluorescence occurs when the rate of electron transfer from the indole ring to an acceptor such as an amide is altered. This could be due to an uneven distribution of charges in the vicinity of the tryptophan where negative charge near the accepting amide and positive charge near the indole can increase the energy required for the transfer of the electron. This will decrease the rate of transfer and thereby decrease the fluorescence quantum yield [56]. There are two tryptophan residues, Trp533 and Trp548 that are in close proximity to residue 540 in FOXP2. In the monomeric form, Trp533 is in close proximity to Glu519 and Arg536. These charged residues could contribute to the quenching of Trp533 fluorescence. Because Arg536 is in the hinge loop region, it is likely that its position could be altered in the Y540F variant which could account for the increased fluorescence of the variant compared to the wild type. Thus structurally, the variant shows both increased backbone order and an alteration to the tertiary structural packing, likely in the environment of the hinge loop.
Although the crystal structure indicates that the wild type protein is capable of forming a domain swapped dimer, this dimer is not detectable at concentrations as high as 100 µM (Fig. 4). The concentration of protein used for the crystal structure was in excess of 600 µM [29]. At concentrations as high as this and under the precipitating conditions needed for crystallisation, dimerisation is promoted. The Y540F variant, however, shows a significant proportion of dimer present in solution at concentrations as low as 30 µM. The mutation therefore increases the dimerisation propensity of the FOXP2 FHD by ~9.5 fold. The reason for this increase can be attributed to the location of residue 540 both in the hinge loop region and at the dimer interface. The increased hydrophobic nature of Phe540 allows it to become more deeply buried at the hydrophobic dimer interface than Tyr540 and it likely forms stabilising contacts with the hydrophobic network of residues at the interface which will increase the stability of the dimer. This is confirmed, in part, by the crystal structures where Phe373 in FOXP3 is in direct van der Waals contact with four hydrophobic residues (Phe340, Leu345, Phe367 and Phe374) while Tyr540 in FOXP2 is only in direct contact with one hydrophobic residue (Phe541). The other residues are present but they are not within van der Waals distance of Tyr540. Phenylalanine at this position will promote the hydrophobic contacts and improve dimerisation. The position of Tyr540 in the hinge loop region is of fundamental significance for domain swapping and the fact that its replacement with a phenylalanine alters backbone structure even in the monomeric form, could suggest that the mutation changes tension in the hinge loop region which would alter susceptibility to form a domain swapped dimer. This, along with the increase in hydrophobicity of the interface could contribute to the increased tendency of the Y540F variant to dimerise.
The residue at position 540 does not only influence dimerisation of the FOXP FHD but it also affects the ability of the protein to bind to DNA. The data in Fig. 5 indicate that both the wild type and the variant are capable of binding DNA. However, it is clear that there are differences in DNA binding between the two.
Firstly, the EMSA results show two convincing trailing bands for the variant but only one for the wild type. These two bands, which are not present under denaturing conditions, are representative of DNA binding to two species of different sizes. This implies that the variant binds DNA both as a monomer and dimer while the wild type only binds as a monomer. This is in accordance with the size exclusion chromatography results which show that at the concentrations used in the EMSA, the variant exists as a mixture of monomer and dimer species while the wild type exists almost exclusively as a monomer. It is interesting to note from these results that both monomer and dimer species are capable of binding DNA.
Secondly, the quantity of DNA binding is reduced in the variant as demonstrated by the lower percentage of total bound DNA despite the fact that the DNA migrates in two separate bands. This implies that the affinity of the FHD for DNA has been reduced by the tyrosine to phenylalanine substitution. The reduced affinity can be appreciated qualitatively by comparing the binding isotherms obtained from the EMSAs. The apparent K d values obtained from these plots suggest that the wild type has a higher affinity for the DNA than the variant (Fig. 5c). The Tyr540-OH forms an H-bond with the DNA backbone according to the crystal structure. This is a water-mediated bond in the case of the monomer and a direct bond in the case of the dimer. The decrease in the affinity upon substitution with a phenylalanine is due to loss of this H-bond with DNA. Thus, although the dimer is stronger when a phenylalanine is at position 540, the affinity of the Y540F variant for DNA decreases. However, due to the higher propensity to form a dimer, the stoichiometry of binding may increase as is the case with FOXP3 where one dimer can simultaneously bind two DNA strands [16]. This has been suggested to be an important physiological role for the dimer where it brings two separate DNA strands into close proximity in order to facilitate the formation of higher order transcriptional complexes. Future work could expand this study by investigating how the mutation affects transcription within cells.
5 Conclusion
The work presented here demonstrates that the conservative substitution of Tyr540 with a phenylalanine in the FOXP2 FHD results in both increased dimerisation propensity and decreased DNA binding affinity. The mutation causes a decrease in disorder of the backbone structure as well as a change in the local environment of certain residues. Dimerisation susceptibility is increased by (1) formation of stabilising hydrophobic contacts at the dimer interface and (2) adjustment of the tension within the hinge loop region. DNA binding affinity is decreased due to the loss of an H-bond with the DNA although DNA binding stoichiometry may increase. Because the strength of dimerisation can vary, because both monomer and dimer are capable of binding to DNA and because the stoichiometry of DNA binding can change, alterations in the monomer dimer equilibrium could be an important mechanism for regulation of transcription by the FOXP subfamily.
Abbreviations
- CD:
-
Circular dichroism
- EMSA:
-
Electrophoretic mobility shift assay
- FHD:
-
Forkhead domain
- FOX:
-
Forkhead box family
- IPEX:
-
Immune dysregulation, polyendocrinopathy, enteropathy and X-linked
References
Kaufmann E, Knochel W (1996) Five years on the wings of fork head. Mech Dev 57(1):3–20
Mazet F, Yu JK, Liberles DA, Holland LZ, Shimeld SM (2003) Phylogenetic relationships of the Fox (Forkhead) gene family in the Bilateria. Gene 316:79–89
Chae WJ, Henegariu O, Lee SK, Bothwell AL (2006) The mutant leucine-zipper domain impairs both dimerization and suppressive function of Foxp3 in T cells. Proc Natl Acad Sci USA 103(25):9631–9636
Su DM, Navarre S, Oh WJ, Condie BG, Manley NR (2003) A domain of Foxn1 required for crosstalk-dependent thymic epithelial cell differentiation. Nat Immunol 4:1128–1135
Friedman J, Kaestner K (2006) The Foxa family of transcription factors in development and metabolism. Cell Mol Life Sci CMLS 63(19–20):2317–2328
Partridge L, Bruning JC (2008) Forkhead transcription factors and ageing. Oncogene 27(16):2351–2363
Lu MM, Li S, Yang H, Morrisey EE (2002) Foxp4: a novel member of the Foxp subfamily of winged-helix genes co-expressed with Foxp1 and Foxp2 in pulmonary and gut tissues. Mech Dev 119(Suppl 1):S197–S202
Rausa FM, Tan Y, Costa RH (2003) Association between hepatocyte nuclear factor 6 (HNF-6) and FoxA2 DNA binding domains stimulates FoxA2 transcriptional activity but inhibits HNF-6 DNA binding. Mol Cell Biol 23(2):437–449
Mecklenburg L, Nakamura M, Sundberg JP, Paus R (2001) The nude mouse skin phenotype: the role of Foxn1 in hair follicle development and cycling. Exp Mol Pathol 71(2):171–178
Castanet M, Park SM, Smith A, Bost M, Leger J, Lyonnet S, Pelet A, Czernichow P, Chatterjee K, Polak M (2002) A novel loss-of-function mutation in TTF-2 is associated with congenital hypothyroidism, thyroid agenesis and cleft palate. Hum Mol Genet 11(17):2051–2059
Saleem RA, Banerjee-Basu S, Berry FB, Baxevanis AD, Walter MA (2003) Structural and functional analyses of disease-causing missense mutations in the forkhead domain of FOXC1. Hum Mol Genet 12(22):2993–3005
Saleem RA, Banerjee-Basu S, Murphy TC, Baxevanis A, Walter MA (2004) Essential structural and functional determinants within the forkhead domain of FOXC1. Nucleic Acids Res 32(14):4182–4193
Hamdan FF, Daoud H, Rochefort D, Piton A, Gauthier J, Langlois M, Foomani G, Dobrzeniecka S, Krebs M-O, Joober R (2010) De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet 87(5):671–678
Vernes SC, Nicod J, Elahi FM, Coventry JA, Kenny N, Coupe A-M, Bird LE, Davies KE, Fisher SE (2006) Functional genetic analysis of mutations implicated in a human speech and language disorder. Hum Mol Genet 15(21):3154–3167
Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD (2001) The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27(1):20–21
Bandukwala HS, Wu Y, Feuerer M, Chen Y, Barboza B, Ghosh S, Stroud JC, Benoist C, Mathis D, Rao A (2011) Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity 34(4):479–491
Banham AH, Beasley N, Campo E, Fernandez PL, Fidler C, Gatter K, Jones M, Mason DY, Prime JE, Trougouboff P, Wood K, Cordell JL (2001) The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res 61(24):8820–8829
Wildin R, Smyk-Pearson S, Filipovich A (2002) Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet 39(8):537–545
Li S, Weidenfeld J, Morrisey EE (2004) Transcriptional and DNA binding activity of the Foxp1/2/4 family is modulated by heterotypic and homotypic protein interactions. Mol Cell Biol 24(2):809–822
Clark KL, Halay ED, Lai E, Burley SK (1993) Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5
van Dongen MJ, Cederberg A, Carlsson P, Enerbäck S, Wikström M (2000) Solution structure and dynamics of the DNA-binding domain of the adipocyte-transcription factor FREAC-11. J Mol Biol 296(2):351–359
Jin C, Marsden I, Chen X, Liao X (1999) Dynamic DNA contacts observed in the NMR structure of winged helix protein-DNA complex. J Mol Biol 289(4):683–690
Tsai KL, Huang CY, Chang CH, Sun YJ, Chuang WJ, Hsiao CD (2006) Crystal structure of the human FOXK1a-DNA complex and its implications on the diverse binding specificity of winged helix/forkhead proteins. J Biol Chem 281(25):17400–17409
Tsai KL, Sun YJ, Huang CY, Yang JY, Hung MC, Hsiao CD (2007) Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res 35(20):6984–6994
Boura E, Rezabkova L, Brynda J, Obsilova V, Obsil T (2010) Structure of the human FOXO4-DBD-DNA complex at 1.9 a resolution reveals new details of FOXO binding to the DNA. Acta Crystallogr Sect D Bioll Crystallogr 66(Pt 12):1351–1357
Littler DR, Alvarez-Fernandez M, Stein A, Hibbert RG, Heidebrecht T, Aloy P, Medema RH, Perrakis A (2010) Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic Acids Res 38(13):4527–4538
Sheng W, Rance M, Liao X (2002) Structure comparison of two conserved HNF-3/fkh proteins HFH-1 and genesis indicates the existence of folding differences in their complexes with a DNA binding sequence. Biochemistry 41(10):3286–3293
Brent MM, Anand R, Marmorstein R (2008) Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure 16(9):1407–1416
Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, Tong H, Chen L (2006) Structure of the forkhead domain of FOXP2 bound to DNA. Structure 14(1):159–166
Schlunegger MP, Bennett MJ, Eisenberg D (1997) Oligomer formation by 3D domain swapping: a model for protein assembly and misassembly. Adv Protein Chem 50:61–122
Liu Y, Gotte G, Libonati M, Eisenberg D (2002) Structures of the two 3D domain-swapped RNase A trimers. Protein Sci Publ Protein Soc 11(2):371–380
Rousseau F, Schymkowitz JW, Itzhaki LS (2003) The unfolding story of three-dimensional domain swapping. Structure 11(3):243–251
Park C, Raines RT (2000) Dimer formation by a “monomeric” protein. Protein Sci Publ Protein Soc 9(10):2026–2033
Janowski R, Kozak M, Jankowska E, Grzonka Z, Grubb A, Abrahamson M, Jaskolski M (2001) Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat Struct Biol 8(4):316–320
Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC (2001) Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol 8(9):770–774
Staniforth RA, Giannini S, Higgins LD, Conroy MJ, Hounslow AM, Jerala R, Craven CJ, Waltho JP (2001) Three-dimensional domain swapping in the folded and molten-globule states of cystatins, an amyloid-forming structural superfamily. EMBO J 20(17):4774–4781
D’Alessio G (1999) The evolutionary transition from monomeric to oligomeric proteins: tools, the environment, hypotheses. Prog Biophys Mol Biol 72(3):271–298
Gotte G, Bertoldi M, Libonati M (1999) Structural versatility of bovine ribonuclease A: distinct conformers of trimeric and tetrameric aggregates of the enzyme. Eur J Biochem FEBS 265(2):680–687
Liu Y, Eisenberg D (2002) 3D domain swapping: as domains continue to swap. Protein Sci Publ Protein Soc 11(6):1285–1299
Newcomer ME (2002) Protein folding and three-dimensional domain swapping: a strained relationship? Curr Opin Struct Biol 12(1):48–53
Cole N, Ralston GB (1994) Enhancement of self-association of human spectrin by polyethylene glycol. Int J Biochem 26(6):799–804
Lindner R, Ralston G (1995) Effects of dextran on the self-association of human spectrin. Biophys Chem 57(1):15–25
Rivas G, Fernandez JA, Minton AP (1999) Direct observation of the self-association of dilute proteins in the presence of inert macromolecules at high concentration via tracer sedimentation equilibrium: theory, experiment, and biological significance. Biochemistry 38(29):9379–9388
Green SM, Gittis AG, Meeker AK, Lattman EE (1995) One-step evolution of a dimer from a monomeric protein. Nat Struct Mol Biol 2(9):746–751
Parker MJ, Dempsey CE, Hosszu LL, Waltho JP, Clarke AR (1998) Topology, sequence evolution and folding dynamics of an immunoglobulin domain. Nat Struct Mol Biol 5(3):194–198
Rousseau F, Schymkowitz JW, Wilkinson HR, Itzhaki LS (2001) Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc Natl Acad Sci USA 98(10):5596–5601
Kuhlman B, O’Neill JW, Kim DE, Zhang KY, Baker D (2001) Conversion of monomeric protein L to an obligate dimer by computational protein design. Proc Natl Acad Sci 98(19):10687–10691
Seeliger MA, Schymkowitz JW, Rousseau F, Wilkinson HR, Itzhaki LS (2002) Folding and association of the human cell cycle regulatory proteins ckshs1 and ckshs2. Biochemistry 41(4):1202–1210
Byeon IJ, Louis JM, Gronenborn AM (2004) A captured folding intermediate involved in dimerization and domain-swapping of GB1. J Mol Biol 340(3):615–625
Frank MK, Dyda F, Dobrodumov A, Gronenborn AM (2002) Core mutations switch monomeric protein GB1 into an intertwined tetramer. Nat Struct Mol Biol 9(11):877–885
Chu YP, Chang CH, Shiu JH, Chang YT, Chen CY, Chuang WJ (2011) Solution structure and backbone dynamics of the DNA-binding domain of FOXP1: insight into its domain swapping and DNA binding. Protein Sci Publ Protein Soc 20(5):908–924
Whitmore L, Wallace BA (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89(5):392–400
Wang B, Lin D, Li C, Tucker P (2003) Multiple domains define the expression and regulatory properties of Foxp1 forkhead transcriptional repressors. J Biol Chem 278(27):24259–24268
Correcirc DH, Ramos CH (2009) The use of circular dichroism spectroscopy to study protein folding, form and function. Afr J Biochem Res 3(5):164–173
Murray AJ, Lewis SJ, Barclay AN, Brady RL (1995) One sequence, two folds: a metastable structure of CD2. Proc Natl Acad Sci 92(16):7337–7341
Callis PR, Liu T (2004) Quantitative prediction of fluorescence quantum yields for tryptophan in proteins. J Phys Chem B 108(14):4248–4259
Larkin MA, Blackshields G, Brown N, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23(21):2947–2948
Acknowledgments
We extend our gratitude to Lin Chen, University of Southern California, Los Angeles, CA for the generous donation of the pET-30 LIC plasmid encoding FOXP2 FHD. This work was supported by the University of the Witwatersrand and the South African National Research Foundation Grants 80681, 89515 and 64788 from the South African Chairs Initiative of the Department of Science and Technology and National Research Foundation. Any opinion, findings and conclusions or recommendations expressed in this material are those of the author(s) and therefore the National Research Foundation, the Department of Science and Technology does not accept any liability with regard thereto.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Perumal, K., Dirr, H.W. & Fanucchi, S. A Single Amino Acid in the Hinge Loop Region of the FOXP Forkhead Domain is Significant for Dimerisation. Protein J 34, 111–121 (2015). https://doi.org/10.1007/s10930-015-9603-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-015-9603-4