
Abstract
The bacterial strain Paenibacillus xylanilyticus KJ-03 was isolated from a sample of soil used for cultivating Amorphophallus konjac. The cellulase gene, cel5A was cloned using fosmid library and expressed in Escherichia coli BL21 (trxB). The cel5A gene consists of a 1,743 bp open reading frame and encodes 581 amino acids of a protein. Cel5A contains N-terminal signal peptide, a catalytic domain of glycosyl hydrolase family 5, and DUF291 domain with unknown function. The recombinant cellulase was purified by Ni-affinity chromatography. The cellulase activity of Cel5A was detected in clear band with a molecular weight of 64 kDa by zymogram active staining. The maximum activity of the purified enzyme was displayed at a temperature of 40 °C and pH 6.0 when carboxymethyl cellulose was used as a substrate. It has 44% of its maximum activity at 70 °C and retained 66% of its original activity at 45 °C for 1 h. The purified cellulase hydrolyzed avicel, CMC, filter paper, xylan, and 4-methylumbelliferyl β-d-cellobiose, but no activity was detected against p-nitrophenyl β-d-glucoside. The end products of the hydrolysis of cellotetraose and cellopentaose by Cel5A were detected by thin layer chromatography, while enzyme did not hydrolyze cellobiose and cellotriose.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Cellulose, the major polysaccharide of plant is one of the most abundant biomass with its estimated synthesis rate of 1010 tonnes per year and the largest renewable carbon source on the earth [10]. Cellulose in plant cell wall consists of a linear polymer of β-1,4-linked glucose residues [1, 15]. Insoluble cellulosic substrates can be converted into useful products such as soluble sugars, alcohol and other compounds by cellulose-degrading microorganisms [2]. The cellulases and cellulolytic enzymes involved in degrading celluloses have been grouped into 118 families based on similarity of amino acid sequence [7]. Especially, cellulases are classified in 16 of the 118 families of glycosyl hydrolases. Cellulases or cellulolytic enzymes are responsible for hydrolytic cleavage of β-1,4-glycosidic bonds in cellulose and be classified into three types based on their mode of action [10]. The endoglucanases or endo-1,4-glucanases randomly hydrolyze the β-1,4 glycosidic bonds in cellulose chains, creating more terminal ends in fragments. Exoglucanases or cellobiohydrolases hydrolyze terminal glycosidic bonds and liberate glucose or cellobiose. Finally, β-glucosidases catalyze the hydrolysis of cellobiose or oligoglucosaccharides [10, 22]. Crystalline and amorphous cellulose are efficiently hydrolyzed by the synergistic action of the above three types of enzymes.
Cellulases can be applied in various industries because they are used in animal nutrition, pulp and paper manufacture, detergent, and bio fuel [3, 9, 26]. In particular, they have been used in the degradation of agricultural wastes and can be converted into soluble sugars, alcohol, and other chemicals. Since converting cellulosic wastes to glucose has a global significance, cellulases play an important role for achieving benefits of biomass utilization [12, 14, 16]. Fermentable sugars are used as a renewable energy source to produce bioethanol. They also improve paper web formation and flexibility in the pulp and paper industry [12].
Although most of microorganisms including bacteria and fungi produce cellulose degrading enzymes, cellulases produced by fungi have considerable interests in bioconversion of renewable lignocelluloses [19, 21]. In fungi, Trichoderma and Aspergillus species are able to produce cellulases and some bacteria such as Bacillus, Chlostridium, Cellulomonas, and Erwinia species have been reported to produce cellulases [8, 20, 24]. Paenibacillus species have been reclassified from Bacillus species based on 16S rDNA sequence in the last decade [27]. Paenibacillus species are capable of producing enzymes that involved in the metabolism of plant carbohydrate polymers because these are isolated and identified from soil and plant related environment [5, 12]. Paenibacillus species are able to secrete various polysaccharides-hydrolyzing enzymes including amylase, cellulase, β-glucanase, pectinase and xylanase [12]. Therefore, Paenibacillus is facultatively aerobic organisms which produce many industrial enzymes and they have potential of utilization in a variety of industries [27]. Recently extensive studies are performed on enzymes of Paenibacillus, but cellulase of P. xylanilyticus has not yet been investigated.
In this study, we reported on the cloning, purification and characterization of cellulase from P. xylanilyticus KJ-03 which has shown hydrolysis efficiency against various polysaccharides.
2 Materials and Methods
2.1 Bacterial Strains and Culture Conditions
Trypic soy broth supplemented 0.1% carboxymethyl cellulose (CMC) was used for the cultivation of P. xylanilyticus KJ-03. Escherichia coli (E. coli) JM109 and E. coli BL21 (trxB) were used as host cells for gene cloning. pUC118 was used as a cloning vector. (Takara, Japan). Recombinant E. coli was cultured in Luria–Bertani broth (LB) supplemented with ampicillin (50 μg/mL) at 37 °C. CMC and cellooligosaccharides were purchased from Sigma chemical. Restriction enzymes and T4 ligase were purchased from Takara (Japan).
2.2 Isolation and Identification of Polysaccharide-Degrading Strain
Strain KJ-03 was isolated from soil samples of Amorphophallus konjac field using tryptic soy agar plate (TSA) supplemented with 0.2% CMC, glucomannan, locust bean gum, chitin, birchwood xylan and starch, respectively. Strains from soil samples were incubated for 24 h at 30 °C and then stained with 0.5% congo red and washed with 1 M NaOH. Strains were stained with KI/I2 solution (2% KI, 0.2% I2) for amylase. The strain KJ-03 formed a large yellow zone around colonies and was finally selected.
The genomic DNA of P. xylanilyticus KJ-03 was prepared by standard method, as described by Sambrook et al. [25]. Chromosomal DNA obtained from KJ-03 was prepared for analysis of 16S rDNA. Polymerase chain reaction was performed to amplify the 16S rDNA coding region using both primers, 5′-GAGTTTGATCCTGGCTCAG-3′ (position 9–27 of E. coli 16S rDNA) and 5′-AGAAAGGAGGTGATCCAGCC-3′ (position 1,542–1,525 of E. coli 16S rDNA). The reaction mixture was subjected to 5 cycles of amplication (60 s at 95 °C, 60 s at 37 °C, 90 s at 72 °C). The cycle was followed by additional 30 cycles of amplication (60 s at 95 °C, 60 s at 55 °C, 90 s at 72 °C) using a thermal cycler (Takara, Japan). The PCR product was cloned into pGEM-T Easy vector (Promega, USA). Nucleotide sequence was determined, and phylogenetic tree was conducted using the ClustalX program.
2.3 Sample Preparation for Scanning Electron Microscope
Sample was pre-fixed by 2% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) with 0.1% MgSO4 for 30 min at 4 °C and centrifuged for removal of pro-fixed buffer. Pre-fixed sample washed 0.1 M cacodylate (pH 7.2) for 1 h in twice and centrifuged for removal. And then pre-fixed sample was fixed by 1% osmium tetroxide in 0.2 M cacodylate buffer (pH 7.4) at 4 °C for 24 h, centrifuged for removal of fixed buffer and washed twice with 0.2 M cacodylate buffer (pH 7.4) for 1 h. Dehydrated sample was done with followed order: 50% ethanol (Et-OH) for 10 min at 4 °C (twice), 70% Et-OH for 10 min at 4 °C (twice), 80% Et-OH for 10 min at 4 °C (twice), 90% Et-OH for 10 min at 4 °C (twice), 95% Et-OH for 10 min at room temperature (twice), and 100% Et-OH for 10 min at room temperature (twice). The dehydrated sample was dried by hexadimethyl-disilazane for 1 h and centrifuged for removal of hexadimethyl-disilazane.
2.4 Construction of Genome Library
A genome library of P. xylanilyticus KJ-03 was constructed using the commercial fosmid vector, pCC1FOS (Epicentre, Madison, WI). Total genomic DNA from P. xylanilyticus KJ-03 was prepared by standard method, as described by Sambrook et al. [25]. The extracted genomic DNA was efficiently sheared by passing it through a pipette tip 100 times. End-repair reaction was performed to generate blunt, 5′-phophorylated DNA. End-repaired DNA was electrophoresed in a 1% low melting point agarose at 35 V for 12 h, and approximately 23–40 kb DNA fragments were extracted from gel using GELase (Epicentre, Madison, WI). The prepared DNA fragments were ligated into the fosmid vector and packaged using a MaxPlax Lamda Packaging Extracts (Epicentre, Madison, WI). The packaged sample was transformed into E. coli EPI300-T1R.
2.5 Subcloning of the Cellulase Gene
The library clones having cellulase activity were selected on LB agar plate supplemented with 0.5% CMC and 12.5 μg/mL chloramphenicol. The clones were incubated for 2 days at 37 °C, stained with 0.5% congo red, and washed with 1 M NaCl. The clones formed a yellow zone around colonies were selected by staining with congo red. For subcloning, fosmid clones were partially digested with Sau3AI. 2–10 kb DNA fragments of the fosmid DNA were ligated into the pUC118/BamHI/CIAP and then transformed into E. coli JM109. A clone (pCMC-1) having cellulase gene formed a yellow zone around colonies on LB agar plate containing 0.5% CMC. The analysis of sequence and database similarity searches were done using the BLAST at National Center for Biotechnology Information (NCBI).
2.6 Expression and Purification of Recombinant Cellulase
The cellulase produced by P. xylanilyticus KJ-03 was expressed by subcloning the gene using the pET-32a(+) (Novagen, Germany). The cellulase was amplified by PCR with CMC-EF-sp (5′-GCAGGATCCGCTCCTGATCACCAAGGC-3′) and CMC-ER (5′-GAGGTCGACTTAAGATGTTTGACCAAC-3′) primers. These primers were modified to contain BamHI and SalI restriction enzyme sites. The 50 μL reaction mixture contained 20 pmol of each primer, 0.1 μg genomic DNA, 20 mM dNTP, and 0.25 U Ex Taq DNA polymerase (TakaRa, Japan), and was subjected to 30 cycles of amplication (30 s at 95 °C, 30 s at 59 °C, and 90 s at 72 °C). The PCR products were digested by BamHI and SalI and ligated into the pET-32a(+). The recombinant plasmid, pET-CMC-sp, was transformed into E. coli BL21 (trxB) for the protein expression.
E. coli BL21 (trxB) harboring pET-CMC-sp was induced for over expression with 50 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an OD600 of 0.5 and cultured again 4 h. Cells were harvested by centrifugation (6,000 rpm for 10 min at 4 °C), and then suspended in binding buffer (20 mM sodium phosphate pH 7.4, 0.1 M NaCl, 5 mM imidazole). The cells were disrupted by sonication and the supernatant was collected by centrifugation (13,000 rpm for 30 min at 4 °C). A HisTrap affinity column (Amersharm Phamacia Biotech.) was equilibrated with binding buffer, and the supernatant was applied to the column. The tagged proteins were eluted with elution buffer (20 mM sodium phosphate, pH 7.4, 0.1 M NaCl, 0.5 M imidazole). The eluted fractions were dialyzed overnight against 20 mM sodium phosphate buffer and concentrated by Amicon Ultra-4 (Millipore, Ireland). The purified fusion proteins were digested with enterokinase for 16 h at 20 °C, and unbound fractions were then collected using a HisTrap affinity column.
2.7 SDS-PAGE and Zymogram
The protein concentrations were determined using Bradford method [4]. Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) was performed by the method of Laemmli [13]. Proteins in the gel were stained with Coomassie Brilliant blue R-250. For detection of cellulase activity, 0.1% CMC was contained in the gel. After electrophoresis, the gel was washed in 25% isopropanol and then soaked in 0.1 M citrate buffer (pH 5.0) for 20 min at 4 °C. The gel was incubated for 1 h at 40 °C, stained with 0.5% congo red for 30 min, and washed with 1 M NaCl.
2.8 Enzyme Assay
The reaction mixture containing 50 μL of 0.1% CMC, 199 μL citrate buffer (pH 6.0), and 1 μL enzyme solution was incubated at 40 °C for 15 min. The amount of amount of reducing sugar was determined by the modified dinitrosalicylic acid (DNS) method using glucose as a standard [17]. One unit of cellulase activity was defined as the amount of enzyme required to produce 1 μmol of reducing sugar per min. The optimal temperature and pH of enzyme was determined by the standard assay at the different temperature (ranging from 20 to 80 °C) and pH conditions (ranging from pH 3 to 10.6). For determination of the optimal pH, 50 mM various buffers, citrate (pH 3–6), sodium phosphate (pH 6–8), Tris–HCl (pH 8–9), and glycine–NaOH (pH 9–10.6) were used. The temperature stability was performed by pre-incubating at different temperatures, 40, 45, and 50 °C without substrates. The pH stability of the enzyme was determined various buffers at 4 °C for 3 h. The remaining activity was measured under the standard assay. The effects of chemical on enzyme activity were assayed using various reagents such as Ca2+, Cu2+, Fe3+, Hg2+, Mg2+, Mn2+, Zn2+, β-mercaptoethanol, and EDTA. Each reagent was added to the enzyme solution at a final concentration of 5 mM.
2.9 Substrate Specificity and Hydrolysis of Cellulose
The substrate specificity of cellulase under the standard condition was determined in reactions with 1% CMC, avicel, Whatman filter paper, birchwood xylan, and 25 mM 4-methylumbelliferyl β-d-cellobiose (4-MUC). 0.025 g filter paper was used to 0.25 mL reaction mixture and reaction mixture was often converted during incubation. The enzyme assays were performed for 2 h at pH 6.0 and 40 °C and the enzyme activities were determined by the DNS method. Reaction with 20 μL 25 Mm 4-MUC, 379 μL 50 mM citrate buffer (pH 6.0), and 1 μL the purified enzyme was incubated at 40 °C for 15 min. The reaction was stop by adding 0.6 ml 1 M Na2CO3 and activity was measured at 410 nm. One unit of activity was defined as the amount of enzyme required to release of 1 μmol of 4-methylumbelliferyl per min. For the hydrolysis products of cello-oligosaccharides, the reaction mixture containing 5 μL of 1% cellooligosaccharides, 5 μL of citrate buffer (pH 6.0) and 1 μL of the purified enzyme was incubated at 40 °C for 2 h. The reaction products of cello-oligosaccharides were analyzed by thin layer chromatography (TLC). Aliquots (1 μL) of the reaction mixtures were analyzed on TLC plates. The plates were developed with n-butanol:acetic acid:water (6:3:2), spayed with aniline–diphenylamine reagent (4 mL of aniline, 4 g of diphenylamine, 200 mL of acetone, and 30 mL of 85% phosphoric acid), and baked at 150 °C for 3 min. Glucose, cellobiose, cellotriose, cellotetraose, and cellopentaose were used as standard oligosaccharides (Sigma, USA).
2.10 GenBank Accession Number
The nucleotide sequences of 16S rDNA and cellulase studied in this work have been assigned as the GenBank accession numbers HQ258920.1 and JN872736, respectively.
3 Results
3.1 Isolation and Identification of Strain KJ-03
Strain KJ-03 was isolated from a sample of soil used for cultivating Amorphophallus konjac by using tryptic soy agar (TSA) containing 0.2% of various polysaccharides. This strain formed a large yellow zone on TSA containing 0.2% CMC, birchwood xylan, oat spelt xylan, glucomannan, and locust bean gum, and a clear zone on TSA containing 0.2% starch. The KJ-03 strain, capable of hydrolyzing various polysaccharides, and was determined to be a gram-positive, rod-shaped bacterium, that was 0.3 μm × 3 μm in size (data not shown). Optimal growth occurred at 28–30 °C, although it also grew at 37 °C. Colonies of KJ-03 on TSA are circular and cream-colored. Partial sequencing of the 16S rDNA indicated that it was most closely related to the genus Paenibacillus, possessing the highest similarity (99%) to the P. xylanilyticus strain YUPP-1. The identities of 16S rDNA were 98% for P. illinoisensis (AB073192), 96% for P. amylolyticus (D85396), 95% for P. pabuli (X60630), 94% for P. favisporus (AY208751) and P. turicensis (AF378694), 92% for P. chibensis (D85395), 91% for P. macquariensis (X57305) and 89% for P. glucanolyticus (D88514), respectively [23] (Fig. 1). The strain was designated as P. xylanilyticus KJ-03.

Phylogenetic tree based on 16S rDNA nucleotides of strain KJ-03 and other Paenibacillus species. The tree is implemented using the neighbor-joining method in ClustalX program. The scale bar indicates 0.1 nucleotide positions
3.2 Cloning of the Cellulase Gene cel5A
Ten of the 2,000 clones capable of hydrolyzing CMC were selected from the KJ-03 genome library. The Fos-CMC-6 DNA fragment was subcloned into pUC118 and the E. coli harboring the recombinant plasmid pCMC-1 formed a yellow zone around the colonies upon Congo red staining for cellulase activity. The pCMC-1 plasmid consisted of 4 open reading frames (ORFs) (Fig. 2a). The predicted protein for ORF1 (370 amino acids) shared 76% amino acid identity with the extracellular solute-binding protein family 1 of Paenibacillus sp. Y412MC10. The predicted proteins for ORF2 (252 amino acids) and ORF3 (312 amino acids) shared 66 and 52% amino acid identity, respectively, with two transcriptional regulator Arac family members and the sensor signal transduction histidine kinase of Paenibacillus sp. Y412MC10. Sequence analysis of the cellulase gene revealed a single ORF of 1,743 bp that encoded a protein of 581 amino acids with a molecular mass of 64 kDa. The N-terminal sequence analysis predicted by SignalP possessed a signal peptide of 35 amino acids (cleavage site before Ala36). Analysis of the deduced amino acid sequence of the cellulase gene showed high similarity with the cellulase belonging to glycosyl hydrolase family 5, and was homologous with the DUF291 domain at the C-terminus of the cellulase gene (Fig. 2b).

Schematic representation of the insert from pCMC-6 clone constructed with predicted four ORFs and the conserved domain of the Cel5A. a The insert from pCMC-6 consisted of four ORFs and ORF4 was appeared as cellulase. b Cel5A has homology to the GH5 cellulase and DUF291 domain, and BglC domain. GH5 cellulase, glycoside hydrolase family 5 cellulase; DUF291, putative lg-like domain; BglC, endoglucanase
3.3 Expression and Purification of Cel5A Fusion Protein in E. coli
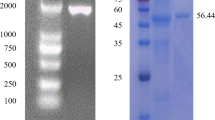
The Cel5A was expressed in E. coli as a fusion protein. The fusion protein was expressed at high levels by induction with 50 mM IPTG, and was purified using Ni-affinity chromatography. SDS-PAGE analysis of the purified enzyme revealed a single band of approximately 64 kDa lacking the signal peptide (Fig. 3). The cellular extract of E. coli harboring Cel5A had a specific activity of 4.8 U/mg, and the enzyme was purified 6.4 folds with a recovery yield of 19%. The purified enzyme showed a strong characteristic band in SDS-PAGE containing 0.1% CMC. These results indicated that the purified enzyme was an apparent cellulase produced by KJ-03.

SDS-PAGE and zymogram of Cel5A. Analysis of the expression of Cel5A and the purified Cel5A were carried in 10% acrylamide gel. Lane 1 protein molcular size markers; lane 2 cell-free lysate; lane 3 purified Cel5A by Ni-affinity chromatography; lane 4 Purified Cel5A digested by Enterokinase; lane 5 zymogram of the purified enzyme for cellulase activity
3.4 Characterization of Cel5A
The effect of pH on the activity of the purified enzyme was investigated in various buffers ranging from pH 3.0 to 10.6 (Fig. 4a). The purified enzyme displayed maximal activity at pH 5.0, with a relative activity of 77% at pH 6.0. In addition, Cel5A maintained up to 40% of its optimal activity at pH 7.0–8.0; however, no activity was observed from pH 9.6 to 10.6. The purified enzyme was stable after it was incubated for 3 h in citrate buffer (pH 6.0). It was acid-labile at pH 6.0 and stable under weak alkaline conditions in the pH range of 6.0–8.0 (Fig. 4b). The purified enzyme was stable from 30 to 70 °C, and sharply decreased to 62% of its original activity after it was incubated in citrate buffer (pH 5.0). The optimal activity of the enzyme was determined to be 40 °C, and more than 65% of the maximal activity was observed at 60 °C (Fig. 5a). Moreover, Cel5A exhibited approximately 40% activity at 20 and 80 °C. To determine the thermal stability of the enzyme, it was incubated at 40, 45, and 50 °C. Cellulase retained 86 and 66% of its activity when incubated at 40 and 45 °C for 1 h. When the incubation temperature was increased to 50 °C, the residual activity decreased dramatically by 74% after 20 min, and remained stable after 1 h incubation period at 40 °C (Fig. 5b). These results indicated that KJ-03 Cel5A has optimal activity at 40 °C and retained activity at high temperatures.

Effect of pH on the activity and stability of cellulase. a The activity of cellulase was assayed at 40 °C and pH ranging from 3.0 to 9.0 with citrate (pH 3–6), sodium phosphate (pH 6–8), and Tris–HCl (pH 8–9). b The pH stability of the enzyme was determined by incubating the enzyme for 3 h at 4 °C in various pHs with citrate (pH 3–6), sodium phosphate (pH 6–8) and Tris–HCl (pH 8)

Effect of temperature on the activity and stability of cellulase. a The activity was assayed at pH 6.0 and temperatures ranging from 20 to 80 °C. b The temperature stability of enzyme was determined by incubating the enzyme for 10–60 min at 30 °C (filled triangle), 40 °C (filled square), and 50 °C (filled circle) in citrate buffer (pH 6.0) without substrate before determining the residual activity of the enzyme
The effect of various metal ions and other reagents on Cel5A activity was studied at 40 °C for 15 min after pre-incubation at 40 °C for 30 min (Table 1). The Ca2+ and Cu2+ ions increased cellulase activity by 38 and 35% of the control, respectively. In contrast, the Fe3+ and Hg2+ ions strongly inhibited, while Zn2+ and Mn2+ decreased its relative activity to 64% and 50%, respectively. The enzyme activity was not affected by Mg2+. EDTA and β-mercaptoethanol inhibited its activity.
3.5 Substrate Specificity and Analysis of Hydrolysis Products
To determine substrate specificity, the purified enzyme was incubated with the substrates CMC, avicel, filter paper, and birchwood xylan. The enzyme showed highest activity against CMC (0.2 U/mg) whereas 64 ± 6.72%, 10 ± 0.11%, and 59 ± 1.89% activity was found on avicel, filter paper, and birchwood xylan, respectively. Hydrolysis was more efficient with CMC than with crystalline cellulose such as avicel and filter paper. The end products of the hydrolysis of cellopentaose by enzyme which cellobiose and cellotriose were major hydrolysis products, and cellotetraose and glucose, minor products were determined by TLC (Fig. 6). It also strongly degraded cellotetraose into cellotriose and glucose, but cellobiose was produced as a minor hydrolysis product. The enzyme did not hydrolyze cellobiose and cellotriose, and was less active on cellotetraose compared with cellopentaose.

Thin layer chromatography analysis of hydrolysis products from cellooligosaccharides by Cel5A. Cel5A was incubated at 40 °C and pH 5.0 with 50 μg cellobiose (lane 2), cellotriose (lane 3), cellotetraose (lane 4), and cellopentaose (lane 5), respectively. Lane 1 contains size marker of glucose (G1), cellobiose (G2), cellotriose (G3), cellotetraose (G4), and cellopentaose (G5); lane 6 Cel5A without cellooligosaccharides
4 Discussion
In this report, we describe the cloning, purification, and characterization of the cel5A cellulase gene from P. xylanilyticus KJ-03 that had been isolated from Konjac field. The deduced amino acids of cel5A contained a catalytic domain for the glycosyl hydrolase family 5 and DUF291 domain. The GH 5 family contained a conserved motif with the consensus region, [LIV]–[LIVMFYWGA](2)–[DNEQG]–[LIVMGST]–{SENR}–N–E–[PV]–[RHDNSTLIVFY]. The cellulase of P. xylanilyticus KJ-03 also conserved the consensus region LMFESINEPR (data not shown). The Glu191 residue in KJ-03 that may play an important role in catalytic reactions mediated by the GH 5 family was conserved in the GH 5 family motif. While the glutamate also acts as a catalytic nucleophile and proton donor in this family. The function of the DUF291 domain is unknown. The enzyme-structure consisted of an Ig-like fold, and DUF291 might mediate interactions with carbohydrates [5]. This analysis revealed a relatively high similarity between this and other cellulase genes in the GH 5 family, including P. polymyxa (67%), P. lautus (65%), Micromonospora sp. (64%), B. halodurans (56%), Dictyoglomus turgidum (35%), and Clostridium thermocellum (32%). The KJ-03 cellulase is secreted into the extracellular media, because a signal peptide sequence exists in the N-terminus of the enzyme. Several members of the Paenibacillus species are capable of producing various polysaccharides that are hydrolyzed by extracellular enzymes such as cellulase, pectinase, xylanase, amylase, and β-glucosidase [12]. Furthermore, Cel5A could degrade polysaccharides such as CMC, xylan, locust bean gum, starch, and chitin, demonstrating its involvement in plant carbohydrate metabolism; it, thus, may be an attractive candidate for bioethanol production and pulp industries.
Compared with some cellulases of P. campinasensis BL11 (pH 7.0), P. polymyxa GS01 (pH 6.0), and B. amyloliquefaciens DL-3 (pH 7.0), Cel5A displayed maximum activity toward CMC at 40 °C and pH 5.0, indicating that it shows optimum activity at lower pH [5, 12, 14]. The optimum activity of Cel5A was at 40 °C, similar to that of Erwinia chrysanthemi PY35 (40 °C) and higher than that of Pseudomonas sp. SK38 (30 °C) [18, 24]. Cellulases produced by Paenibacillus sp. such as P. polymyxa GS01, Paenibacillus sp. strain B39, and P. campinasensis BL11 are optimally active from 50 to 60 °C, whereas the optimal temperature of Cel5A was found to be lower than that of other Paenibacillus sp. cellulases [5, 12, 27]. Equal concentration (5 mM) of Ca2+ and Cu2+ ions stimulated KJ-03 cellulase activity. Similarly, the endoglucanase of Paenibacillus sp. strain B39 and P. campinasensis BL11 in GH family 5 showed a slight increase in activity with Ca2+ [12, 27]. These results indicated that Ca2+ is important for catalytic activity. Since Hg2+ might bind enzyme-amino acid thiol groups, tryptophan residues, or carboxyl groups, the cellulase activity was inhibited by Hg2, while EDTA decreased enzyme activity, indicating that EDTA removes metal ions for enzyme activation. The reducing agent β-mercaptoethanol slightly decreased cellulase activity, suggesting that disulfide bonds are essential to maintain of the enzyme-structure.
The KJ-03 cellulase hydrolyzed CMC more efficiently than the insoluble cellulosic substrates such as avicel and filter paper, and displayed higher activity toward CMC than avicel or filter paper, which was similar to the cellulase characteristics of other GH 5 family members including P. campinasensis BL11, Paenibacillus sp. strain B39, and Vibrio sp. G21 [5, 6, 12]. In addition, Cel5A hydrolyzed crystalline cellulose (such as avicel and filter paper), as well as birchwood xylan. In particular, plant cells are insoluble, and therefore, the highly soluble cellulose, CMC, is widely used as substrate for studies involving endoglucanases [16]. The use of CMC for cellulase activity suggests the involvement of weak cellulolytic enzymes, since most organisms that cannot hydrolyze cellulose can degrade CMC. The KJ-03 cellulase can degrade insoluble cellulose (avicel) at 64% relative activity, and therefore, it can be utilized to produce soluble sugars from insoluble cellulose during bioethanol production. In addition, Cel5A degraded cellotetraose and cellopentaose, but not cellobiose and cellotriose. Other cellulases generally yield cellobiose and cellotriose as the final products from cellotetraose; however, Cel5A mainly produced cellotriose as the final product, with little cellobiose. The hydrolysis products obtained from cellobiose and cellotriose by KJ-03 cellulase-mediated hydrolysis were similar to those of CelAM11 and Cel5A of E. cellulosolvens [11, 28]. CelAM11 and Cel5A of E. cellulosolvens preferentially hydrolyzed cellotetraose to cellobiose, whereas Cel5A of KJ-03 mainly produced cellotriose and glucose. These results indicated that patterns of the purified enzyme on TLC were related to endoglucanase activity, but differed from other cellulases [6, 11, 28].
In conclusion, we have cloned and characterized a cellulose gene from P. xylanilyticus KJ-03, which belongs to the GH 5 family. Considering the ability of the enzyme to function over a broad range of pH and temperature, KJ-03 cellulase has the potential to be used in industrial applications. Cellulases are used for the production of bioethanol from lignocellulosic biomass. This enzyme can degrade cellulosic materials from lignocellulosic biomass and generate fermentable sugars. Therefore, recombinant Cel5A may be valuable for increasing the efficiency of sugar and bioethanol production.
Abbreviations
- Cel5A:
-
Cellulase from Paenibacillus xylanilyticus KJ-03
- LB:
-
Luria–Bertani
- TSA:
-
Tryptic soy agar
- CMC:
-
Carboxymethyl cellulose
- IPTG:
-
Isopropyl-β-d-thiogalactopyranoside
- SDS-PAGE:
-
Sodium dodecyl sulphate polyacrylamide gel electrophoresis
- DNS:
-
Dinitrosalicylic acid
- DTT:
-
Dithiothreitol
- EDTA:
-
Ethylenediaminetetraacetic acid
- TLC:
-
Thin layer chromatography
- ORF:
-
Open reading frame
- GH 5 family:
-
Glycosyl hydrolase 5 family
References
Akita M, Kayatama K, Hatada Y, Ito S, Horikoshi K (2005) FEMS Microbiol Lett 248:9–15
Bayer EA, Chanzy H, Lamed R, Shoham Y (1998) Curr Opin Struct Biol 8:548–557
Bayer EA, Lamed R, Himmel ME (2007) Curr Opin Biotechnol 18:237–245
Bradfod MM (1976) Anal Biochem 72:248–254
Cho KM, Hong SJ, Math RK, Asraful Islam SM, Kim JO, Lee YH, Kim H, Yun HD (2008) J Basic Microbiol 48:464–472
Gao Z, Ruan L, Chen X, Zhang Y, Xu X (2010) Appl Microbiol Biotechnol 87:1373–1382
Henrissat B (1991) Biochem J 280:309–316
Hansen CK, Diderichsen B, Jorgensen PL (1992) J Bacteriol 174:3522–3531
Ito S (1997) Extremophiles 1:61–66
Jung YJ, Lee YS, Park IH, Chandra MS, Kim KK, Choi YL (2010) Electron J Biotechnol 47:203–210
Kim DW, Kim SN, Baik S, Park SC, Lim CH, Kim JO, Shin TS, Oh MJ, Seong CN (2011) J Microbiol 49:141–145
Ko CH, Tsai CH, Lin PH, Chang KC, Tu J, Wang YN, Yang CY (2010) Bioresour Technol 101:7882–7888
Laemmli UK (1970) Nature 227:680–685
Lee YJ, Kim BK, Lee BH, Jo KI, Lee NK, Chung CH, Lee YC, Lee JW (2008) Bioresour Technol 99:378–386
Lima AOS, Quecine MC, Fungaro MHP, Andreote FD, Macche-roni W Jr, Araujo WL, Silva Filho MC, Pizzirani-Kleiner AA, Azevedo JL (2005) Appl Microbiol Biotechnol 68:57–65
Lynd LR, Weimer PJ, Zyl WHV, Pretorius IS (2002) Microbiol Mol Biol Rev 66:506–577
Miller L (1959) Anal Chem 31:208–218
Park SR, Kim MK, Kim JO, Cho SJ, Cho YU, Yun HD (1999) Mol Cells 10:269–274
Pason P, Kyu KL, Ratanakhanokchai K (2006) Appl Environ Microbiol 72:2483–2490
Paster FIJ, Pujol X, Blanco A, Vidal T, Torres AL, Diaz P (2001) Appl Microbiol Biotechnol 55:61–68
Pereira JH, Chen Z, McAndrew RP, Sapra R, Chhabra SR, Sale KL, Simmons BA, Adams PD (2010) J Struct Biol 172:372–379
Posta K, Beki E, Wilson DB, Kukolya J, Hornok L (2004) J Basic Microbiol 5:383–399
Rivas R, Mateos PF, Molina EM, Velazquez E (2005) Int J Syst Evol Microbiol 55:405–408
Ryu SK, Cho SJ, Park SR, Lim WJ, Kim MK, Hong SY, Bae DW, Park YW, Kim BK, Kim H, Yun HD (2001) Appl Microbiol Biotechnol 57:138–145
Sambrook J, Russel DW (2001) Molecular cloning a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Wang F, Li F, Chen G, Liu W (2009) Microbiol Res 164:650–657
Wang CM, Shyu CL, Ho SP, Chiou SH (2008) Lett Appl Microbiol 47:46–53
Yoda K, Toyoda A, Mukoyama Y, Naakanura Y, Minato H (2005) Appl Environ Microbiol 71:5787–5793
Acknowledgment
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2011-0008619).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Park, IH., Chang, J., Lee, YS. et al. Gene Cloning of Endoglucanase Cel5A from Cellulose-Degrading Paenibacillus xylanilyticus KJ-03 and Purification and Characterization of the Recombinant Enzyme. Protein J 31, 238–245 (2012). https://doi.org/10.1007/s10930-012-9396-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-012-9396-7