
Abstract
Second-generation antisense oligonucleotides (ASOs) demonstrate excellent biological stability and in vitro/in vivo potency, and thus are considered to be attractive candidates for drugs to treat various diseases. A pharmacokinetic–pharmacodynamic (PK–PD) model of ASOs is desired for the design of appropriate PK and pharmacological studies. The objective of this study was to develop a PK–PD model to accurately simulate hepatic ASO concentration and its efficacy from plasma ASO concentration. After single subcutaneous administration of an ASO targeting hepatic apolipoprotein B (Apo-B) mRNA to mice, the ASO was absorbed rapidly and showed biphasic decline with time from the plasma and liver (t1/2: 1–3 and 81–183 h, Tmax: 0.25–0.50 and 4–8 h). After administration, hepatic Apo-B mRNA and plasma total cholesterol began decreasing at 4–8 and 8–24 h, and their Tmax values were observed at 24–72 and 72 h. To develop the PK–PD model based on the mechanisms of ASOs, we described the plasma and hepatic ASO concentration with linear two-compartment models. In addition, we inserted two indirect response models for mRNA and plasma total cholesterol. Model predictions from plasma ASO concentration gave excellent fits to the observed values of hepatic ASO concentration, Apo-B mRNA and plasma total cholesterol after single or multiple subcutaneous administrations. Our PK–PD model could accurately predict hepatic ASO concentrations and their efficacies from plasma ASO concentrations. This PK–PD model could be a useful tool for suggesting PK and pharmacological study protocols for various liver-targeted second-generation ASOs.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Antisense oligonucleotides (ASOs) are short synthetic single-strand nucleotide polymers (typically of 7–30 nucleotides in length) that are designed to specifically hybridize to their target RNA via Watson–Crick base pairing and to prevent expression of the encoded “disease-related” protein product [1]. In particular, second-generation ASOs with protective nucleotides, such as locked nucleic acid (LNA) [2], 2′O-methoxyethyl [3] and amino bridged nucleic acid [4], have led to dramatic advances for biological stability, which consequently provided longer half-lives in target tissues compared to conventional ASOs. Second-generation ASOs have a gapmer structure which is designed with a central DNA segment (6–10 nucleotides) that is flanked at each molecular terminus by 2–5 protective nucleotides. The gapmer structure efficiently induces the cleavage of the target RNA by recruiting endogenous ribonuclease H (RNase H) as a major pathway to translational inhibition [5, 6], resulting in potent pharmacological actions with fewer side-effects.
Following subcutaneous injection of second-generation ASOs, they are rapidly and well absorbed from a subcutaneous injection site [1, 7]. After reaching the Cmax, the plasma concentration shows multi-phasic decline with a fast initial distribution phase that dominates the plasma exposure (AUC0–24 h or AUC0–48 h represent 90 % or more of total AUC), and a slow terminal elimination phase that consists of very low plasma concentration. Little or no accumulation of Cmax and total AUC is observed by repeated administrations. After systemic circulation, second-generation ASOs are almost completely taken up by tissues such as the liver and kidney via endocytosis. In the tissues, second-generation ASOs are slowly metabolized by nucleases and as a consequence, remain for a long time. Second-generation ASOs and their metabolites are ultimately eliminated via excretion primarily in urine. The pharmacokinetics of second-generation ASOs appear to be very similar and independent of sequence, facilitating potentially more rapid clinical drug development compared to low-molecular compounds. Clinical trials of second-generation ASOs are on-going for the treatment of various diseases such as hyperlipidemia [8] and cancer [9].
While there have been some reports on the pharmacokinetic–pharmacodynamic (PK–PD) analysis of second-generation ASOs, most of them have only been on the relationship between the ASO concentration and mRNA reduction in the target tissue [5, 10]. Only a few studies have tried modeling the in vivo PK–PD relationship of ASO. Yu et al. reported for the first time, an in vivo PK–PD model for ASO with an indirect response model assuming that ASO would induce degradation of the target RNA by stimulation of RNase H, and could subsequently predict reduction of the target RNA from the ASO concentration of hepatocytes after single or multiple administration [11]. However, the ASO concentration of hepatocytes that they used was not precisely quantitated, and their model did not include the plasma ASO concentration and the therapeutic biomarker response, which are clinically available. Callies et al. reported a PK–PD model for human ASO exposure and therapeutic biomarker response from preclinical PK and pharmacological data [12]. They could predict the plasma ASO concentration, but not the tumor ASO concentration, target RNA and protein in human because the model that was integrated from preclinical PK and pharmacological data could not be physiologically interlocked, and its estimated parameters were not relevant physiological values in human. For drug development, it is important to include the plasma ASO concentration and the therapeutic biomarker response, which are clinically available, into the model for accurate prediction of tissue ASO concentration, target RNA and therapeutic biomarker response.
Straaup et al. reported that LNA gapmers of ASO designed to target the hepatic apolipoprotein B (Apo-B) mRNA, which is important for cholesterol metabolism, remained in the liver for a long time and improved the plasma total cholesterol level via reduction of Apo-B mRNA in mice after administration [5]. In this investigation, we used this ASO as a model compound for second-generation ASOs and measured the plasma and hepatic ASO concentration, hepatic Apo-B mRNA, and plasma total cholesterol after single or multiple subcutaneous administration to mice. The objectives of this study were to develop a PK–PD model for ASO based on the mechanism of ASO, and to accurately simulate the hepatic ASO concentration and its efficacy from the plasma ASO concentration.
Methods
Chemicals
The ASO was purchased from GeneDesign, Inc. (Osaka, Japan). The sequence of the ASO is 5′-GCattggtatTCA-3′ (gapmer oligonucleotide with LNA (capital) and DNA (plain font);. all internucleoside linkages are phosphorothioated), which has the highest affinity and potency for Apo-B mRNA [5]. Saline was purchased from Otsuka Pharmaceuticals (Tokyo, Japan). RNAlater was purchased from QIAGEN, (Hilden, Germany). TE buffer (pH 8.0) was obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). All other reagents and solvents were commercial products of reagent grade.
Animals
Animal care and all experimental procedures were performed with the approval of the Institutional Animal Care and Use Committee of Shionogi in terms of the 3R (replacement/reduction/refinement) principle. Female C57BL/6J Jcl mice were purchased at 7 weeks of age from CLEA Japan Inc. (Osaka, Japan). After quarantine for a week, the mice were acclimated for several days in the animal compartment. The mice were used for the experiments at 8 weeks of age (body weight 18–20 g). During the acclimation and experimental periods, the mice were placed under the conditions of room temperature of 20–26 °C, relative humidity of 30–70 %, and lighting for 12 h (light [8:00–20:00]/dark [20:00–8:00]) and allowed free access to tap water and solid laboratory food (CE-2, CLEA Japan, Inc.).
Animal experiments
ASO was dissolved in saline which was used as the control and subcutaneously administered to mice (n = 3/time point) at single (1, 5 and 10 mg/kg) or multiple (5 mg/kg/week, 4 weeks) doses. For determination of plasma ASO concentration, blood samples were collected via the inferior vena cava with a syringe attached to a needle containing heparin and EDTA-2K under isoflurane anesthesia at 0.083, 0.25, 1, 4, 8 and 24 h. For the determination of plasma total cholesterol concentration, they were collected at 0, 1, 4, 8, 24, 72, 168, 336, 504 and 672 h. After blood collection, liver samples were collected for determination of hepatic ASO concentration at 0.083, 0.25, 1, 4, 8, 24, 72, 168, 336, 504 and 672 h or for determination of Apo-B mRNA at 0, 1, 4, 8, 24, 72, 168, 336, 504 and 672 h. In the multiple administration study, blood and liver samples were collected at 168, 336, 504 and 672 h. The blood samples were centrifuged at 1,600 g for 10 min at 4 °C. The isolated plasma and liver samples were stored frozen at −80 °C until analysis.
Determination of plasma and hepatic concentration of ASO, hepatic Apo-B mRNA and plasma total cholesterol
The concentrations of ASO in plasma and liver samples were determined by liquid chromatography/tandem mass spectrometry (LC–MS/MS) using API5000 (AB SCIEX, Foster City, CA, USA) by a previously reported procedure with slight modifications [13]. In brief, liver samples were homogenized in extraction buffer (0.5 % Nonidet P-40, 25 mM ethylenediaminetetraacetic acid, 100 mM sodium chloride, 25 mM tris hydroxymethyl aminomethane, 0.5 mg/mL proteinase K, pH 8.0) and incubated at 37 °C for 3 h. ASO in plasma and liver homogenate samples was extracted by a liquid–liquid extraction method using phenol/chloroform (1/1, v/v), followed by solid-phase extraction (Oasis HLB, 10 mg, Waters Inc., Milford, MA, USA). The eluent was evaporated to dryness under nitrogen and reconstituted in 10 μM H3PO4 in TE buffer (pH 8.0)/methanol (9/1, v/v). After the reconstituted solution was filtrated through a 0.22 µm pore size filter, 5 µL of filtrated solution was injected into the LC–MS/MS system. Chromatographic separations were performed at a flow rate of 0.2 mL/min on ACQUITY BEH C18 (2.1 mm × 50 mm, 1.7 µm, Waters Inc., Milford, MA, USA) for 8 min. A binary gradient was used to perform the separations. Mobile phase A consisted of 400 mM HFIP and 15 mM TEA in water, and mobile phase B consisted of mobile phase A/methanol/acetonitrile (2/1/1). The column temperature was maintained at 60 °C using a column heater. The range of calibration standards were from 0.03 to 30 μg/mL for plasma and from 0.03 to 30 μg/g for liver.
Total RNA was extracted from liver homogenates using RNeasy 96 Universal Tissue Kit (QIAGEN). One μg of total RNA was converted to cDNA using SuperScript™ III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen, Camarillo, CA, USA), according to the manufacturer’s instructions. Quantitative PCR was performed with SYBR® Premix Ex Taq II (Takara Bio, Otsu, Japan) using Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Apo-B mRNA levels were normalized to GAPDH and are presented relative to the saline control.
Plasma total cholesterol concentration was determined by the colorimetric method with T-cholesterol E-test Wako kits (Wako Pure Chemical Industries, Ltd.).
Non-compartment analysis
Pharmacokinetic analysis of the averaged plasma and hepatic concentration per time was performed by WinNonlin Professional (ver 5.0.1, Pharsight Inc.) based on a non-compartment model with uniform weighting. The area under the plasma and hepatic concentration–time curve (AUC) was calculated by the trapezoidal rule. The elimination half-life (t1/2) was calculated by linear regression using logarithmic values of appropriate points. The other pharmacokinetic parameters were as follows: maximum plasma and hepatic concentration (Cmax), time to reach maximum plasma and hepatic concentration (Tmax).
PK–PD modeling
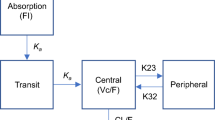
Figure 1 presents a PK–PD model for ASO. As the plasma concentration of second-generation ASOs shows multi-phasic decline [1], the plasma concentration of ASO was described by a linear two-compartment open model as follows:
where Asc, Apla and Aper are the ASO amount (mg/kg) in the subcutaneous, plasma and peripheral compartment; Vdp is the distribution volume of ASO in the plasma compartment (L/kg); ka and kep are the first-order rate constants for absorption from the injection site and elimination from plasma compartment (/h); k12,P and k21,P are the first-order rate constants for plasma to peripheral and peripheral to plasma compartment (/h). Cpla is the plasma ASO concentration (μg/mL).

PK–PD model for the reduction of hepatic Apo-B mRNA and plasma total cholesterol by ASO
In the modeling of the hepatic behavior of ASO, the “sub-compartment” was incorporated into the proposed PK model to accurately calculate the hepatic ASO concentrations at the terminal phase. Therefore, a linear two-compartment model was used as follows:
where Aliv and Asub are the ASO amount (mg/kg) in the liver and sub-compartment; VdL is the distribution volume of ASO in the liver compartment (kg of live/kg of body weight); F is the distribution ratio of ASO from the plasma compartment to the liver compartment; k12,L and k21,L are the first-order rate constants from the liver to the sub-compartment and from the sub-compartment to the liver compartment (/h); keL is the first-order rate constant (/h) for elimination from liver compartment; and Cliv is the ASO concentration (μg/g of liver) in the liver compartment. Due to the lack of sensitivity of LC–MS/MS, we could not adequately characterize the elimination phase in the plasma and liver.
In the pharmacodynamic model structure, the change in the hepatic Apo-B mRNA expression was determined by a zero-order rate constant (kin,mRNA, % of control/h) for the synthesis of mRNA and a first-order rate constant (kout,mRNA,/h) for the degradation of mRNA. The drug-effect relationship between ASO amount in the sub-compartment and kout,mRNA could be described by an indirect response model as follows:
where RmRNA is the mRNA expression (% of control) in the Apo-B mRNA compartment; SmaxmRNA is the maximum reducing effect of mRNA (% of control); and SC50mRNA is the ASO concentration in the sub-compartment (mg/kg) reducing 50 % of SmaxmRNA. RmRNA,0 shows the basal level of Apo-B mRNA (% of control) and RmRNA,ss is the maximum potency of Apo-B mRNA reduction at steady state. In the indirect response model, the SmaxmRNA is determined by Eq. (10). To minimize the number of parameters to be estimated, SmaxmRNA was fixed as 100 in the modeling. The basal level of RmRNA before ASO administration was maintained as RmRNA,0, which was determined as \({\text{k}}_{{{\text{in}},{\text{mRNA}}}} = {\text{ k}}_{{{\text{out}},{\text{mRNA}}}} \cdot {\text{R}}_{{{\text{mRNA}},0}}\).
As Apo-B mRNA causes production of very low-density lipoprotein (VLDL) in the liver, an additional indirect response compartment was inserted. In this model structure, the change of VLDL in the compartment and plasma cholesterol concentration was demonstrated as follows:
where RVLDL is the value (% of control) of the VLDL in the VLDL compartment; RVLDL,0 is the basal level (% of control) of VLDL; ImaxCho is the maximum hypocholesterol effect (mg/dL) of the VLDL; IC50Cho is the RVLDL (% of control) reducing 50 % of ImaxCho; Echo,0 is the basal level (mg/dL) of plasma total cholesterol; and γ is the Hill function. The change in the VLDL was determined by a zero-order rate constant (kin,VLDL, % of control/h) for the synthesis of VLDL and a first-order rate constant (kout,VLDL,/h) for the degradation of VLDL. The basal level of RVLDL before ASO administration was maintained as RVLDL,0, which was determined as \({\text{k}}_{{{\text{in}},{\text{VLDL}}}} = {\text{ k}}_{{{\text{out}},{\text{VLDL}}}} \cdot {\text{R}}_{{{\text{VLDL}},0}}\).
Data analysis
ASO concentrations in plasma and liver after subcutaneous administration of ASO to mice represent the mean ± standard deviation (SD) as shown in Supplemental Table 1. All parameters were estimated by fitting the PK–PD models to the average of the observed data at each time point using WinNonlin Professional althouh the elimination phase in the plasma and liver could not be adequately characterized. We selected normal distribution error as the error model and sequential approach. Correlation analysis for the observed versus predicted values was also performed to determine the goodness of fit for the model compared to lines of identity (y = x). Repeated simulations were carried with the PK–PD model by NONMEM (ver 7.2.0) because WinNonlin Professional could not simulate the repeated dose expected for built-in models.
Results
Pharmacokinetic analysis for plasma and hepatic ASO concentration
Figure 2 shows the plasma and hepatic ASO concentrations after subcutaneous administration of ASO at 1, 5 and 10 mg/kg. ASO was rapidly absorbed and showed biphasic decline with time from plasma. The ASO of plasma could not be detected at 24 h after administration at all dosing. The plasma t1/2 values after administration at 1, 5 and 10 mg/kg were 1.15, 3.05 and 2.46 h, and Tmax values were 0.25, 0.50 and 0.25 h (Table 1). Hepatic ASO concentration showed biphasic decline with time, and hepatic t1/2 values were 81.2, 183 and 173 h, and Tmax were 8, 8 and 4 h. Since linearity was shown by AUC, and slow elimination at late time points was observed, we selected linear two-compartment models to describe the plasma and hepatic ASO concentration (Eqs. 1–7). The model appeared to provide a reasonable fit of the observed concentrations with reasonable variability in parameter estimation although the coefficient of variation (CV) values of k12,P and k21,P were slightly large (Fig. 2 and Table 2).

ASO concentrations in plasma (a) and liver (b) after subcutaneous administration of ASO. Each observed value represents the mean ± SD (n = 3) at 1 mg/kg (diamonds), 5 mg/kg (triangles) and 10 mg/kg (squares) and each predicted value represents the line at 1 mg/kg (solid), 5 mg/kg (dashed) and 10 mg/kg (gray)
PK–PD analysis for hepatic Apo-B mRNA and plasma total cholesterol
Hepatic Apo-B mRNA began decreasing between 4 and 8 h after administration in a dose-dependent manner. The time of the maximum reduction level for hepatic Apo-B mRNA expression ranged from 24 to 72 h. In fact, there was a time lag between hepatic ASO concentration and Apo-B mRNA expression level, and a typical counter-clockwise hysteresis was observed (data not shown). To describe the time-course of the hepatic Apo-B mRNA, an indirect response model was applied to the PK–PD model as shown in Fig. 1 and Eqs. 8–10. As a result, the optimized model nicely predicted the Apo-B mRNA expression as shown in Fig. 3a, even at an early phase of the reduction (Fig. 3b).

Changes in hepatic Apo-B mRNA (a, b) and plasma total cholesterol (c, d) after administration of ASO. Each observed value represents the mean ± SD (n = 3) at control (circles), 1 mg/kg (diamonds), 5 mg/kg (triangles) and 10 mg/kg (squares) and each predicted value represents the line at control (dotted 0 h), 1 mg/kg (solid), 5 mg/kg (dashed) and 10 mg/kg (gray). b, d Shows a magnification for the early phase of a, c
As a therapeutic biomarker response of Apo-B mRNA reduction, plasma total cholesterol concentration was monitored in mice following subcutaneous administration of ASO at 1, 5 and 10 mg/kg. For the dosing of 1 mg/kg, the concentration was less potently reduced with a maximal 20 % decrease. However, stronger reduction (60–80 % at maximal level) was observed after the administration of 5 and 10 mg/kg, and this was retained over 672 h. Plasma total cholesterol concentration showed the maximum reduction at 72 h, which was a slightly later response compared with Apo-B mRNA (Tmax = 24–72 h). To describe the time-course of the plasma total cholesterol concentration, an additional indirect response model including the VLDL compartment was applied to the PK–PD model as shown in Fig. 1 and Eqs. 11–12. The optimized model nicely predicted the plasma total cholesterol concentration as shown in Fig. 3c, even in an early phase of the reduction (Fig. 3d). These data showed that the PK–PD model could describe both the Apo-B mRNA and plasma total cholesterol reduction from the plasma ASO concentration.
PK–PD model evaluation
The correlation analysis between the observed and predicted values for plasma ASO concentration, hepatic ASO concentration, hepatic Apo-mRNA, and plasma total cholesterol was visualized (Fig. 4). These results indicated that the predicted value corresponds well to the observed one.

Correlation analysis for the observed and predicted plasma ASO concentration (a), hepatic ASO concentration (b), hepatic Apo-B mRNA (c), and plasma total cholesterol (d). The dashed lines are lines of identity (y = x)
Subsequently, we simulated the time-course of hepatic ASO concentration, hepatic Apo-B mRNA and plasma total cholesterol after repeated subcutaneous administration of ASO at 5 mg/kg/week for 4 weeks using estimated PK and PD parameters (Tables 2, 3) for external validation of the model. The solid lines shown in Fig. 5 represented the model-simulated profiles of hepatic ASO concentration,Apo-B mRNA expression level and plasma total cholesterol level. Simulated trough values for each index were quite close to the original data. Consequently, we were able to validate the present PK–PD model.

Comparison of simulated values (solid line) with the observed values (triangles) of hepatic ASO concentration, Apo-B mRNA expression and plasma total cholesterol concentration after repeated subcutaneous administration of ASO (5 mg/kg/week). Each observed value represents the mean ± SD (n = 3) at control (circles) and 5 mg/kg (triangles). Dotted line shows control (at 0 h)
Discussion
In the present study, we developed a PK–PD model to characterize hepatic ASO concentration, hepatic Apo-B mRNA expression level and plasma total cholesterol level from plasma ASO concentration. In order to validate the model, we evaluated its simulation of pharmacological activities following multiple dosing of ASO. There were two key points in the development of this model: (i) description of the plasma and hepatic ASO concentrations using linear two-compartment models and (ii) insertion of two indirect response models to describe the reduction of mRNA and plasma total cholesterol based on the pharmacological mechanism of ASO.
In general, following subcutaneous injection of second-generation ASOs, they are rapidly and well absorbed from the subcutaneous injection site, and after reaching the Cmax, the plasma concentration shows a multi-phasic decline with a fast initial distribution phase and a slow terminal elimination phase [1, 7]. Most of the ASOs are taken up by tissues such as the liver and kidney from the systemic circulation. In this study, the ASO after subcutaneous administration showed biphasic decline from the plasma, and quickly distributed to the liver (Fig. 2), as reported previously [11]. The plasma and hepatic ASO concentrations could be analyzed with two-compartment models, and the model appeared to provide a reasonable fit of the observed concentrations. However, the CV values of k12,P and k21,P, which were first-order rate constants between plasma and peripheral compartments, were slightly large because the terminal elimination phase of plasma at 24 h could not be adequately evaluated due to limitation of the analytical method (the lower limit of quantification of LC–MS/MS: 30 ng/mL). As a method for analyzing ASO, hybridization ELISA has very high sensitivity (sub-ng/mL level) and can evaluate the terminal elimination phase of plasma, but it can not distinguish full-length ASO (unchanged form) from the metabolites, resulting in cross-hybridization and overestimation of the unchanged ASO concentration [14, 15]. In this study, we used LC–MS/MS because of its high sensitivity (ng/mL level) and selectivity. We considered it to be appropriate for the PK–PD modeling of unchanged ASO in the plasma and liver. However, at present, LC–MS/MS can not detect sub-ng/mL levels [13, 15, 16]. We also could not characterize the equilibrium between plasma and liver although Yu et al. suggested that the plasma concentration of ASOs in the terminal phase represents the equilibrium with the liver [10]. We could not also characterize the elimination from the plasma compartment. The predicted values with our PK–PD model correspond well to the observed ones (Fig. 4 and 5), suggesting that deleting these parameters has only a small impact on this analysis. Therefore, additional studies are required to evaluate the elimination phase of plasma, the equilibrium between plasma and liver and the elimination from the plasma compartment in order to adequately understand the PK–PD model when the sensitivity of LC–MS/MS can be improved.
We described the amount of ASO in the sub-compartment for the promotion of mRNA degradation. Regarding the mechanism of intracellular trafficking of ASO, there are the two distinct pathways of the productive uptake pathway and the non-productive uptake pathway [17, 18]. The productive uptake pathway, which delivers ASO to the target mRNA cellular component (nucleus), physiologically means that part of the ASO after cellular uptake escapes from endosomes via endocytosis and enters the nucleus, while the non-productive uptake pathway, which does not deliver ASO to the target mRNA, physiologically means that other ASO after cellular uptake move to the lysosome via endocytosis and are eventually degraded. We represented the sub-compartment as the productive uptake pathway to enter the nucleus, hybridize mRNA and promote mRNA degradation by RNase H. The non-productive uptake pathway was considered to be a degradation pathway in the liver, and included part of the elimination (keL) from the liver compartment.
The distribution volume of ASO in the liver compartment was represented as VdL/F, which was the value of the liver volume adjusted for the distribution ratio of ASO from the plasma compartment to the liver compartment. We represented it as VdL/F because the value of VdL adjusted by F (approximately 10 % of the dosing in this study) was similar to that of the physiological liver volume of the mouse (0.065 L/kg) [19], and the bioavailability of second-generation ASOs after subcutaneous administration is generally very good [1].
Regarding the pharmacological mechanism of ASO, hepatic Apo-B mRNA expression was well described by an indirect response model (Fig. 3a, b). Two mechanisms are known for translational inhibition of ASO: (i) induction of cleavage of the target RNA by RNase H and (ii) blockage of the translational apparatus [20]. In this study, we used the LNA gapmer which induces cleavage of the target RNA by recruiting RNase H as a major pathway to translational inhibition [5, 6]. We incorporated only the former into the model and described the indirect response model, as did previous reports [11, 12]. The predicted profiles could also be described precisely at an early phase (Fig. 3b).
With respect to the plasma total cholesterol profiles, it was necessary to insert an additional compartment, the VLDL compartment, in order to describe the time lag between Apo-B mRNA and plasma cholesterol concentration with an indirect response model (Fig. 3c, d). As Apo-B is known to causes production of VLDL in the liver, its reduction suppresses VLDL production, and the reduction of VLDL subsequently causes the reduction of LDL and eventually affects the reduction of the plasma total cholesterol level. Therefore, the currently obtained time lag for the Apo-B mRNA expression and plasma cholesterol concentration might indicate such processing events to form total cholesterol in plasma. We considered this PD model to be more intuitive although we did not measure the profiles of VLDL and LDL after ASO administration. By introducing the VLDL compartment, the time-course of plasma total cholesterol could also be described precisely at an early phase (Fig. 3d). The IC50cho value was estimated to be 73 %, indicating that the maximum effect of the reduction does not lead to zero plasma total cholesterol because reduction of Apo-B mRNA expression does not affect high-density lipoprotein, which constitutes part of the plasma total cholesterol [5]. The Hill function was estimated to be 3.6 (Table 3), indicating that plasma total cholesterol increases steeply with a change in the hepatic Apo-B mRNA expression. Such pharmacometric approaches would be useful for understanding the pharmacological mechanism of ASO and designing an effective pharmacological study. Thus, we were able to analyze hepatic Apo-B mRNA and the plasma total cholesterol by inserting appropriate compartments of the pharmacological mechanism. On the other hand, some disparities between the observed and simulated values were observed in the plasma total cholesterol after administration at 10 mg/kg. Such disparities may be reduced by using detailed analysis of LDL because reduction of Apo-B directly suppresses the production of VLDL and LDL. In addition, we consider that an appropriate value for Imaxcho may be determined by detailed analysis of LDL although we have expedientially fixed the Imaxcho at 64.9 mg/dL which is the basal level of control. In order to accurately predict its efficacy and set the appropriate value of Imaxcho, additional studies are required to develop the PK–PD model based on detailed analysis of cholesterol.
Yu et al. reported an in vivo PK–PD model for ASO with the indirect response model assuming that ASO induced the degradation of the target RNA by stimulation of RNase H, and could subsequently predict the reduction of the target RNA from the ASO concentration of hepatocytes after single or multiple administration [11]. They described a two-compartment model of hepatocytes and non-parenchymal cells in the liver. The ASO concentration of hepatocytes could not be precisely quantitated because ASOs preferentially accumulate in non-parenchymal cells, and the concentration was affected by the dosing amount and time after dosing [11, 21]. On the other hand, we described a liver two-compartment model with sub-compartments for intracellular trafficking and developmentally added the plasma ASO concentration and the therapeutic biomarker response, which are clinically available, into the model for accurate prediction of the hepatic ASO concentration and better efficacy. Callies et al. reported a PK–PD model for human ASO exposure and therapeutic biomarker response from preclinical PK and pharmacological data [12]. They could predict the plasma ASO concentration in human. However, they could not predict the tumor ASO concentration, target RNA and protein in human because the model integrated from preclinical PK and pharmacological data could not be physiologically interlocked, and its estimated parameters of the model were not relevant physiological values in human. In addition, anti-tumor efficacy of human is different from that of the preclinical tumor model. We developed the PK–PD model using the physiologically interlocked PK and pharmacological data. However, our model has only been applied to the mouse and remains untested in non-rodents and humans. In order to accelerate translational research for ASOs, additional studies are required to evaluate the feasibility of this PK–PD model for chimeric mice with humanized liver, other animal species and humans.
In the estimation of pharmacokinetic parameters, ka was relatively large (8.5/h). The ka of second-generation ASOs in mice would be relatively large because the ASOs are as water-soluble and metabolically stable as antibiotics, and the ka of antibiotics in mice was relatively large (20.5/h) [22]. The keP was approximately 72 times larger than keL; this was reasonable because the t1/2 of the liver calculated by non-compartment model analysis was 60–71 times larger than that of the plasma. In addition, our PK–PD model was developed based on reasonable data because Straarup et al. reported the t1/2 (9–14 days) of liver and the hepatic ASO concentrations at 24 h after administration (1, 5 and 10 mg/kg: approximately 0.7, 5 and 8 µg/g), and these values were comparable to those of our study. In the estimation of pharmacodynamic parameters, the value (17.3 mg/kg) of SC50mRNA was converted into 1.5 µg/g of liver adjusted by the physiological liver weight of the mouse (87.5 g of liver/kg of body weight) [19]. It was similar to the value (<10 µg/g liver) of hepatic ASO concentration reducing 50 % of Apo-B mRNA [5]. The value (10.3 h) of the half-life calculated by kout,mRNA was comparable to the value (16 h) of half-life for degradation of Apo-B mRNA [23]. These findings suggest the reasonableness of the SC50mRNA and kout,mRNA calculations.
After repeated administration of ASO, the simulated values of our PK–PD model showed a close fit with the observed values (Fig. 5). Our PK–PD model could capture the initial distribution phase following administration, as above stated. In general, the plasma exposure of the initial distribution phase dominates the total plasma exposure, and little or no accumulation of Cmax and total AUC is observed by repeated administrations [1]. Therefore, except for the terminal elimination phase following administration, our PK–PD model could be applied to multiple dose conditions and be useful for designing various types of pharmacological studies. This PK–PD model might be adaptable to various liver-targeted ASOs because the pharmacokinetics and pharmacological mechanism of the second-generation ASOs appear to be very similar [1, 7].
In conclusion, we successfully developed a PK–PD model to predict the hepatic ASO concentration and its efficacies from plasma ASO concentration in mice. To develop a PK–PD model based on the mechanisms of ASOs, we described the plasma and hepatic ASO concentration with linear two-compartment models. In addition, we inserted two indirect response models for mRNA and plasma total cholesterol. This PK–PD model could be a useful tool for suggesting PK and pharmacological study protocols for various liver-targeted second-generation ASOs.
References
Yu RZ, Grundy JS, Geary RS (2013) Clinical pharmacokinetics of second generation antisense oligonucleotides. Expert Opin Drug Metab Toxicol 9:169–182. doi:10.1517/17425255.2013.737320
Grünweller A, Hartmann RK (2007) Locked nucleic acid oligonucleotides: the next generation of antisense agents? BioDrugs 21:235–243
Kimber WL, Puri N, Borgmeyer C, Ritter D, Sharov A, Seidman M, Ko MS (2003) Efficacy of 2-methoxyethoxy-modified antisense oligonucleotides for the study of mouse preimplantation development. Reprod Biomed Online 6:318–322
Obika S, Rahman SM, Song B, Onoda M, Koizumi M, Morita K, Imanishi T (2008) Synthesis and properties of 3′-amino-2′,4′-BNA, a bridged nucleic acid with a N3′ → P5′ phosphoramidate linkage. Bioorg Med Chem 16:9230–9237. doi:10.1016/j.bmc.2008.09.013
Straarup EM, Fisker N, Hedtjärn M, Lindholm MW, Rosenbohm C, Aarup V, Hansen HF, Ørum H, Hansen JB, Koch T (2010) Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res 38:7100–7111. doi:10.1093/nar/gkq457
Grünweller A, Wyszko E, Bieber B, Jahnel R, Erdmann VA, Kurreck J (2003) Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res 31:3185–3193
Geary RS (2009) Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol 5:381–391
Thomas GS, Cromwell WC, Ali S, Chin W, Flaim JD, Davidson M (2013) Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol 62:2178–2184. doi:10.1016/j.jacc.2013.07.081
Chi KN, Siu LL, Hirte H, Hotte SJ, Knox J, Kollmansberger C, Gleave M, Guns E, Powers J, Walsh W, Tu D, Eisenhauer E (2008) A phase I study of OGX-011, a 2′-methoxyethyl phosphorothioate antisense to clusterin, in combination with docetaxel in patients with advanced cancer. Clin Cancer Res 14(3):833–839
Yu RZ, Lemonidis KM, Graham MJ, Matson JE, Crooke RM, Tribble DL, Wedel MK, Levin AA, Geary RS (2009) Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem Pharmacol 77:910–919. doi:10.1016/j.bcp.2008.11.005
Yu RZ, Zhang H, Geary RS, Graham M, Masarjian L, Lemonidis K, Crooke R, Dean NM, Levin AA (2001) Pharmacokinetics and pharmacodynamics of an antisense phosphorothioate oligonucleotide targeting Fas mRNA in mice. J Pharmacol Exp Ther 296:388–395
Callies S, André V, Patel B, Waters D, Francis P, Burgess M, Lahn M (2011) Integrated analysis of preclinical data to support the design of the first in man study of LY2181308, a second generation antisense oligonucleotide. Br J Clin Pharmacol 71:416–428
Turnpenny P, Rawal J, Schardt T, Lamoratta S, Mueller H, Weber M, Brady K (2011) Quantitation of locked nucleic acid antisense oligonucleotides in mouse tissue using a liquid–liquid extraction LC–MS/MS analytical approach. Bioanalysis 3:1911–1921. doi:10.4155/bio.11.100
Yu RZ, Geary RS, Levin AA (2004) Application of novel quantitative bioanalytical methods for pharmacokinetic and pharmacokinetic/pharmacodynamic assessments of antisense oligonucleotides. Curr Opin Drug Discov Devel 7:195–203
Cen Y, Li X, Liu D, Pan F, Cai Y, Li B, Peng W, Wu C, Jiang W, Zhou H (2012) Development and validation of LC–MS/MS method for the detection and quantification of CpG oligonucleotides 107 (CpG ODN107) and its metabolites in mice plasma. J Pharm Biomed Anal 70:447–455
Ewles M, Goodwin L, Schneider A, Rothhammer-Hampl T (2014) Quantification of oligonucleotides by LC–MS/MS: the challenges of quantifying a phosphorothioate oligonucleotide and multiple metabolites. Bioanalysis 6:447–464
Geary RS, Wancewicz E, Matson J, Pearce M, Siwkowski A, Swayze E, Bennett CF (2009) Effect of dose and plasma concentration on liver uptake and pharmacologic activity of a 2′-methoxyethyl modified chimeric antisense oligonucleotide targeting PTEN. Biochem Pharmacol 78:284–291
Koller E, Vincent TM, Chappell A, De S, Manoharan M, Bennett CF (2011) Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res 39:4795–4807
Davies B, Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095
Lee RG, Crosby J, Baker BF, Graham MJ, Crooke RM (2013) Antisense technology: an emerging platform for cardiovascular disease therapeutics. J Cardiovasc Transl Res 6:969–980. doi:10.1007/s12265-013-9495-7
Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, Schmidt K, Zhao C, Aghajan M, Murray HF, Riney S, Booten SL, Murray SF, Gaus H, Crosby J, Lima WF, Guo S, Monia BP, Swayze EE, Seth PP (2014) Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res 42:8796–8807
Dandekar PK, Tessier PR, Williams P, Nightingale CH, Nicolau DP (2003) Pharmacodynamic profile of daptomycin against Enterococcus species and methicillin-resistant Staphylococcus aureus in a murine thigh infection model. J Antimicrob Chemother 52(3):405–411
Pullinger CR, North JD, Teng BB, Rifici VA, Ronhild de Brito AE, Scott J (1989) The apolipoprotein B gene is constitutively expressed in HepG2 cells: regulation of secretion by oleic acid, albumin, and insulin, and measurement of the mRNA half-life. J Lipid Res 30(7):1065–1077
Acknowledgments
The authors would like to thank Ms. Keiko Ogawa, Mr. Takanori Hironaka, Dr. Tatsuya Ikehara, Dr. Ken-ichi Nezasa, Mr. Shingo Sakamoto, and Dr. Shuichi Onishi (Drug Metabolism and Pharmacokinetics, Drug Developmental Research Laboratories, Shionogi & Co., Ltd.) for their support during and discussion related to the animal experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Shimizu, R., Kitade, M., Kobayashi, T. et al. Pharmacokinetic–pharmacodynamic modeling for reduction of hepatic apolipoprotein B mRNA and plasma total cholesterol after administration of antisense oligonucleotide in mice. J Pharmacokinet Pharmacodyn 42, 67–77 (2015). https://doi.org/10.1007/s10928-014-9398-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-014-9398-5