
Abstract
The PK/PD of abatacept, a selective T cell co-stimulation modulator, was examined in rats with collagen-induced arthritis (CIA) using a nonlinear mixed effect modeling approach. Male Lewis rats underwent collagen induction to produce rheumatoid arthritis. Two single-dose groups received either 10 mg/kg intravenous (IV) or 20 mg/kg subcutaneous (SC) abatacept, and one multiple-dose group received one 20 mg/kg SC abatacept dose and four additional 10 mg/kg SC doses. Effects on disease progression (DIS) were measured by paw swelling. Plasma concentrations of abatacept were assayed by enzyme-linked immunosorbent assay. The PK/PD data were sequentially fitted using NONMEM VI. Goodness-of-fit was assessed by objective functions and visual inspection of diagnostic plots. The PK of abatacept followed a two-compartment model with linear elimination. For SC doses, short-term zero-order absorption was assumed with F = 59.2 %. The disease progression component was an indirect response model with a time-dependent change in paw edema production rate constant (k in ) that was inhibited by abatacept. Variation in the PK data could be explained by inter-individual variability in clearance and central compartment volume (V 1 ), while the large variability of the PD data may be the result of paw edema production (k 0 in ) and loss rate constant (k out ). Abatacept has modest effects on paw swelling in CIA rats. The PK/PD profiles were well described by the proposed model and allowed evaluation of inter-individual variability on drug- and DIS-related parameters.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The etiology and pathology of rheumatoid arthritis (RA) are not well understood. Several environmental and genetic factors, such as tobacco smoking, certain HLA-DR (human leukocyte antigen class II molecules) and PTPN22 (protein tyrosine phosphatase, non-receptor type 22) risk alleles, have been found to be strongly associated with RA [1]. Since the HLA class II molecules are important in presenting antigens to CD4+ T cells, RA is thought to be caused by certain arthritogenic antigen(s) [2]. Currently, no specific antigen for RA has been identified, although several possible endogenous antigens have been discovered. These include antigens that are present in the joint (type 2 collagen and chondrocyte glycoprotein gp39), and ubiquitous antigens such as glucose-6-phosphate isomerase [3]. Some exogenous agents, such as bacterial or viral proteins, have been investigated as well [4].
RA presumably starts with T cell activation, which requires an antigen-specific signal and a co-stimulatory signal [5]. The first signal involves the recognition of arthritogenic antigen by antigen-presenting cells (B cells, macrophages, or dendritic cells), which then bind to CD4+ T-cells through the interaction between T-cell receptor (TCR) and MHC complex. Another signal essential for complete T-cell activation is by the binding of a co-stimulatory receptor on T cell and a ligand on antigen-presenting cells. The best characterized signals are interactions between CD28 on CD4+ T cells and CD80 (B7-1) or CD86 (B7-2) on antigen-presenting cells [6].
Abatacept (CTLA-4Ig) is a soluble fusion protein that contains the Fc region of human immunoglobulin G1 (IgG1) and human cytotoxic T-lymphocyte antigen (CTLA)-4. It is the first member of the co-stimulation blockers [7]. CTLA-4 (also known as CD152) is naturally expressed on the surface of T cells and it competitively inhibits binding between CD28 and CD80/CD86, thereby suppressing T cell activation. Although it is very effective in inhibiting the co-stimulatory signal (binding efficiency to CD80/CD86 is 20-fold higher than CD28), its natural expression is very low compared with CD28 and only becomes detectable after TCR recognizes the MHC complex [8]. With the use of abatacept, T-cell activation is not complete, thus immune responses are suppressed. Previous clinical and pre-clinical studies had shown that abatacept can decrease the expression of cytokines and other biomarkers such as rheumatoid factor (RF) and C-reactive protein [9].
Abatacept (brand name: Orencia) was developed by Bristol-Myers Squibb (BMS) and was first approved for treatment of RA and juvenile idiopathic arthritis in 2005 [10]. It was initially formulated to be administered as a 30-min IV infusion every 2–4 weeks and can be used either as monotherapy or concomitantly with other disease-modifying anti-rheumatic drugs such as methotrexate (MTX) [9]. In 2011, weekly SC dosing of abatacept was also approved, providing more convenience to patients [9]. Although abatacept has demonstrated clinical success in RA treatment and produces chronic improvement of physical function in patients [9], detailed information about its mechanisms of action is unknown. In our study, we aimed to investigate the effects of abatacept on RA by the use of a well-established CIA rat model.
Our laboratory has published a mechanistic disease progression (PK/PD/DIS) model to describe the inter-regulation of glucocorticoids and inflammatory cytokines (interleukin (IL)-1, IL-6 and tumor necrosis factor (TNF)-α) in RA and the PD effects (on paw edema and bone mineral density) of dexamethasone (DEX) in CIA Lewis rats [11, 12]. We have also investigated the PK/PD/DIS relationships of therapeutic proteins (etanercept and anakinra) using CIA rats [13, 14]. The current study was enacted to seek better understanding of the pharmacology of abatacept with quantitative assessment of its PK/PD properties.
Materials and methods
Drug
Abatacept [250 mg lyophilized powder per vial, BMS (Princeton, NJ, USA)] was purchased from a local pharmacy. Abatacept was reconstituted with 10 mL blank Lewis rat plasma. The vial was gently swirled for complete dissolution, and then the reconstitution was diluted with sterile saline for proper concentration before injection. Abatacept injection solution was freshly prepared with a new drug vial on every injection day.
Animals
Fifty male Lewis rats (6–9 weeks old) were purchased from Harlan (Indianapolis, IN, USA) with weights of 150–175 g. Animals were housed individually in the University Laboratory Animal Facility and acclimatized for 1 week under constant temperature (22 °C), humidity (72 %), and 12-h light/12-h dark cycle. Rats had free access to rat chow and water. All protocols followed the Principles of Laboratory Animal Care (Institute of Laboratory Animal Resources, 1996) and were approved by the University at Buffalo Institutional Animal Care and Use Committee.
Induction of collagen-induced arthritis (CIA) in Lewis rats
The induction of CIA in Lewis rats followed protocols and used reagents supplied by Chondrex, Inc. (Redmond, WA, USA). The detailed procedures of the collagen induction have been previously described [11, 13].
Experimental design
Paw edema and body weights were monitored throughout the entire study starting from day 0. Edema was indicated by swelling of the rat hind paws and was measured with digital calipers (VWR Scientific, Rochester, NY, USA) as previously described [11–15]. Edema was indicated by the sum of the paw and ankle area measurements for each hind foot. After evaluation of paw edema on day 20 post-induction, 23 CIA rats with paw volume increases of at least 50 % in one or two paws were selected and randomly assigned to four groups: vehicle control group which received either single (n = 3) or multiple (n = 3) blank SC injections, IV group (n = 5) which received 10 mg/kg IV abatacept, SC single-dose group (n = 6) which received 20 mg/kg SC abatacept, and SC Multiple-dose group (n = 6) which received 20 mg/kg SC abatacept on day 21 with subsequent 10 mg/kg SC doses on days 23, 25, 27, and 29. Control or single-dose groups received injections on day 21.
Serial blood samples (for PK assessment) were collected from the saphenous vein, using EDTA as an anti-coagulant. Blood samples for the IV group were taken at post-dose days 0.0417 (1 h), 1, 2 4, 8, and 14, while for the SC Single-dose group time points were at days 0.125 (3 h), 0.333 (8 h), 1, 2, 4, 6, 8, 12, and 14. For the SC Multiple-dose group, samples were withdrawn at days 0.125, 2, 4, 6, 8, 8.5, 9, 10, 12, and 14 after the first dose at day 21. Blood was collected right before dosing for the SC Multiple-dose rats on the injection days. All blood samples were centrifuged at 2,000×g for 10 min at 4 °C. Plasma fractions were immediately transferred into micro-centrifuge tubes on ice and stored frozen at −80 °C before further analysis.
Enzyme-linked immunosorbent assay (ELISA) methodology
Plasma abatacept concentrations were determined by ELISA. Human sCTLA-4 Platinum ELISA kit was purchased from eBioscience, Inc. (San Diego, CA, USA). All samples were run in duplicate. The overall procedures followed the manufacturer’s instructions except for the following modifications: All samples were diluted with 1× phosphate buffered saline before adding to the wells. No color-giving reagents in the kit were used. The linear range of the assay was 0.16–10 ng/mL. Two quality control samples (2.5 and 0.5 ng/mL) were included for assessing precision and accuracy. The lower limit of quantification of the assay was 0.16 ng/mL and the inter-day variability was around 15.0 %.
Pharmacokinetic model
Compartmental models were used to describe abatacept pharmacokinetics in CIA rats. One-, two-, and three-compartment PK models were tested to fit the IV data. Several models were tried to capture the SC absorption kinetics, including first-order absorption (Bateman function), short-term infusion, and a 2-step approach (zero-order infusion followed by a first-order process). The equations in this section depict the final model selected.
The model for the 10 mg/kg IV group was a mammillary two-compartment model with linear elimination from the central compartment:
where A 1 and A 2 are amounts of drug in the central (V 1 ) and peripheral (V 2 ) compartments, CL and CL D are elimination and distribution clearances (CL), and C P = A 1 /V 1 . For SC injection, a short-term zero-order input was used for the absorption process.
where F is bioavailability and τ is the input time period. The plasma concentration–time profiles of the three treatment groups were fitted simultaneously, and the parameter estimates were then fixed and applied in the combined PK/PD model. Model residual variability was described with a combined additive and proportional model.
Pharmacodynamics and disease progression model
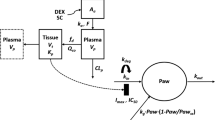
Two models were compared for fitting paw edema disease progression. The first model consisted of a model parameter (t onset ) for describing the lag time observed before the rapid raise of paw edema and a subsequent feedback function (R deg ) on the production of paw edema. This model was previously applied for describing the effects of etanercept [13] in CIA rats. The other was a transduction-based feedback model that contained a series of transit compartments which contributed both to the production and the natural edema remission. This model functioned well for characterizing the effects of anakinra in CIA rats [14]. The temporal changes of paw sizes for the three abatacept dosing groups were fitted simultaneously. The equations for the selected model shown in Fig. 1 were as follows:

where Paw is the sum of ankle and paw size of the rat hind foot. Only arthritic paw measurements were included in the PD analysis. The k grow is a zero-order rate constant accounting for natural paw growth, and t onset is a time delay in disease onset after collagen induction. The zero-order production rate k in is a function of time starting at the onset time, and R deg represents a linear decline in k in and accounts for the natural remission of arthritis after disease onset. The first-order rate constant k out describes the loss of edema. Drug-related parameters include the capacity constant I max (maximum loss of paw edema caused by abatacept) and sensitivity constant IC 50 (abatacept concentration producing 50 % of maximum inhibition).
Model fitting and data analysis
The PD effects of abatacept were assessed by comparison of area under the effect curve (AUEC). Post-dose paw edema data of each rat paw was normalized with their respective value at the beginning of drug administration (on day 21), and the AUEC was calculated between day 21 till the end of the study (day 47). AUEC of paw edema profiles were statistically tested by the two-sided Bailer-Satterthwaite method [16, 17], and the t-statistic (tobs) was calculated as:
where AUEC C and AUEC D are the AUECs of the control and drug-treated rat paw edema, and s 2(AUEC) is the variance of the AUEC. The paw edema profiles were considered statistically different if tobs ≥ 2.39, which is the Bonferroni-corrected critical value (zcrit) in this analysis.
Model fittings were performed by the first-order conditional estimation method with the interaction module in NONMEM VI [18]. The PK profiles were first fitted and the resulting parameter estimates were fixed and applied in the combined PK/PD model. The inter-animal variability of the parameters was modeled with an exponential function. The residual variability model was selected among additive, proportional, and combined additive and proportional models. The number of fixed and random effect parameters was determined by χ2 discrimination test, where a change of 3.84 in the objective function reflects a significant effect of a parameter. The final model was evaluated based on the value of the objective function, precision of parameter estimates, visual inspection of various diagnostic plots, and visual predictive check plots. Visual predictive checks and diagnostic plots were constructed using the R program [19]. Model simulations were conducted with the Berkeley Madonna (Version 8.3.18) software (Berkeley Madonna Inc., Berkeley, CA, USA).
Results
Pharmacokinetics
Figure 2 depicts the concentration–time profiles of the three treatment groups. The abatacept concentration–time profile after IV dosing showed a poly-phasic decline. Abatacept concentration rose rapidly soon after SC Single-dosing and remained flat for 2–4 days, and then declined mono-exponentially. The trough concentrations after SC multiple-doses gradually increased during the treatment period and peaked 2 days after dosing terminated (on day 29). The terminal phases of all three dosing groups were parallel, suggesting unchanged elimination kinetics across the three groups. Noncompartmental (NCA) analysis suggested that abatacept exhibits linear PK across the studied doses. The NCA CL is 20.8 mL/day/kg, volume (V ss ) is 146 mL/kg, and bioavailability (F) of the SC dose dosing is 57.7 %.

Plasma abatacept concentration versus time profiles after a 10 mg/kg IV (squares, n = 5), b SC single-dose 20 mg/kg (triangles, n = 6), and c SC multiple-dose (20 mg/kg with 4 more 10 mg/kg doses every 2 days) (circles, n = 6) administration in CIA rats. Solid lines and dash lines depict median and 90 % confidence interval of model predictions. Note that day 0 reflects day 21 post-induction
Figure 1 shows the structure of the selected PK/PD model. A two-compartment model with linear elimination best fitted the IV PK profile. For SC doses, a short-term infusion type of absorption pattern best described the absorption kinetics. The 2-step absorption model (zero-order infusion followed by a first-order process) also captured the data, but it resulted in a higher objective function and less accuracy in parameter estimates. Figure 2 shows the observed PK data and 90 % confidence interval (CI) of the model predictions. All data points fell within the 90 % CI of the model predictions, which indicated that the PK model well captured the concentration–time profiles of the three dosing groups. Figure 3 displays the diagnostic plots for the PK model. The weighted residuals are distributed evenly across the zero line (Fig. 3a, b) and the individual predictions improved after the random effects were considered (Fig. 3c, d).

Model diagnostic plots of abatacept PK model in CIA rats showing a weighted residuals versus population predictions, b weighted residuals versus post-dose time, c population model predictions versus observed concentrations and d individual model predictions versus observed concentrations. Lines are the zero lines (in a and b) or the identity lines (in c and d)
The PK parameter estimates are listed in Table 1. Overall, model parameters were estimated with good precision. The estimated bioavailability of the SC dose was 59.2 %, which is close to 57.7 % as calculated by NCA and a previous reported value of 62.5 % [20]. The zero-order input duration was estimated to be 2.67 days. Inter-individual variability appeared to be significant for both CL and V 1 parameters, which accounted for 9.67 and 56.2 % of the variability in PK data.
Pharmacodynamics and disease progression
Disease progression of paw volume in both control and abatacept-treated CIA rats and the PD model predictions are shown in Fig. 4 with typical patterns of delay, rise to peak, and later fall off. Statistical analyses were performed to compare the AUEC of the time curves of paw size ratio changes normalized with respective initial values. The normalization was necessary since CIA rats exhibited variable degrees of responses to collagen induction and abatacept treatment. The AUEC (mean ± SE) for the control, IV, SC single-dose, and SC multiple-dose groups before abatacept administration (on day 21) were 24.0 ± 0.2, 23.3 ± 0.3, 22.9 ± 0.4, and 23.5 ± 0.3 fold × day and are not significantly different from each other. After the paw edema data was normalized with the value on day 21, the AUEC of the SC single-dose (20.0 ± 0.3 fold × day) and the SC multiple-dose (19.9 ± 0.3 fold × day) groups were significantly lower (tobs = 3.25 and 3.81, respectively) than the control group (21.2 ± 0.2 fold × day), reflecting the effects of abatacept on reducing paw edema. The SC multiple-dose group showed significantly greater (tobs = 2.50) paw edema reduction compared with the IV dose group (20.8 ± 0.2 fold × day). No differences were found in the other pairwise comparisons.

Time courses of paw volume after dosing with: a vehicle control (closed circles, n = 6), b 10 mg/kg IV (squares, n = 5), c 20 mg/kg SC (triangles, n = 6), and d multiple SC (20 mg/kg with 4 more 10 mg/kg doses every 2 days) (open circles, n = 6) administration at day 21 post-induction in CIA rats. Solid and dashed lines depict median and 90 % confidence interval of model predictions
Both the simple model and the transduction-based model well described the time delay and later profile of the paw edema disease progression. However, the simpler model was selected since it required fewer parameters, provided better precision of parameter estimates, and resulted in a lower objective function value. Figure 4 depicts the observed paw edema data and the model fittings (90 % CI) of the four groups. The model provides reasonable characterization of the paw edema for the control and drug dosing groups, with a slight over-prediction of the IV dose group. Figure 5 shows the goodness-of-fit plots of the PD model. The residuals distribute symmetrically around the zero line, and there is good agreement between the observations and model predictions.

Model diagnostic plots of abatacept PD model in CIA rats showing a weighted residuals versus population predictions, b weighted residuals versus post-induction time, c population model predictions versus observed paw edema; and d individual model predictions versus observed paw edema. Lines are the zero lines (in a and b) or the identity lines (in c and d)
Table 2 provides the PD parameter estimates. The only parameter that was not estimated is R deg , which was fixed to the previously reported value of 0.128/day [13]. All parameters were estimated with acceptable precision, with a slightly higher relative standard error (RSE % of 131.7 %) for IC 50. The t onset parameter was 14.3 days, which is close to the 14.5 days value found in the etanercept study [13]. The maximum effect of abatacept on paw edema (I max ) was 0.161, which agrees with its modest effects. The IC 50 of abatacept was 0.731 μmol/L. Considering the inter-individual variabilities in k grow , k out , k 0 in , and Paw 0 was necessary to generate the lowest objective function. They were estimated to account for 63.1, 6.40, 18.1, and 4.93 % of the variability in the PD data. No improvements were found when the inter-individual variabilities of the drug-related parameters (I max and IC 50) were incorporated.
Discussion
Pharmacokinetics
Abatacept PK have been investigated in different species [20–26]. All these studies indicated that abatacept exhibits linear kinetics following single or multiple doses. Our analysis also agreed with these findings. The use of a two-compartment model has been reported for abatacept. Population analysis of human abatacept PK data also indicated the same model worked well for data from clinical trials [25, 26]. All PK parameter estimates were consistent with the values previously reported by BMS in rats [20]. Our CL was 21.8 mL/day/kg, close to 28.1 mL/day/kg reported. The V ss reported was 149 mL/kg, similar to our 131 mL/kg as the sum of V 1 (69.5 mL/kg) and V 2 (61.9 mL/kg) (Table 1). Variability in our rat PK data can be explained by the inter-individual variability in both CL and V 1 , which is consistent with the findings in human studies [25]. These parameters are typically proportional to body weight in adult animals and humans. The inter-individual variability in CL may also result from the differences in glomerular filtration rate, which was found to be an important covariate for CL in a previous population analysis of human abatacept PK [25].
The SC bioavailability of abatacept (dosages 10–200 mg/kg) in rats ranged between 41.1 and 62.5 %, but tended to decrease as doses increased [20]. Our model estimated bioavailability was 59.2 %. Two different bioavailability terms (F) were tested for the two dose levels in our analysis, but did not decrease the objective function value. Therefore, only one F parameter was included in the final model. The bioavailability of SC dosing in humans is 78.6 % [9]. The interspecies difference in SC bioavailability may be explained by factors such as product formulation, skin morphology, proteolytic activity, or injection site [27]. In our study, SC abatacept solution was prepared using lyophilized drug reconstituted with rat plasma, whereas a prefilled syringe containing abatacept solution was injected in humans. Another possibility is immunogenicity, which may occur in rats due to the humanized nature of abatacept. Although not measured in our study, the formation of anti-abatacept antibodies has been observed in other animal studies [20, 21, 23, 24]. It has been found previously that more than 33 % of the rats receiving SC multiple doses of abatacept showed immunogenic responses [20]. This percentage is much higher than the <1 % found in a clinical trial [28].
Our study showed that abatacept absorption kinetics following SC injection in CIA rats was slow, which agrees with the previous findings in rats and mice [20, 23]. The absorption pattern can be described as a short-term constant input, and the duration was estimated to last for 2.67 days. It is difficult to compare the SC absorption pattern in humans and rats because of the absence of literature reports on the details of human SC PK. However, it is reported that in humans SC abatacept not only has a similar PK pattern with IV infusion, but also exhibits a comparable safety and efficacy profile and low immunogenicity [28]. The absorption pattern for these two administration routes also appeared to be similar for other macromolecules such as trastuzumab and golimumab [29, 30]. These observations suggested the suitability of mimicking the SC absorption process using constant input.
Pharmacodynamics and disease progression
The CIA rat model is the most frequently used experimental model to mimic the human disease with an advantage of a relatively short time-frame and readily measured disease markers [11–15]. This CIA model shares many similar disease characteristics with human RA, such as synovial immune infiltration, pannus formation and joint destruction, and it also exhibits many common immunological factors seen in human RA, including RF and anti-collagen type II antibodies. However, it is not completely identical to RA, and some differences include the much longer latent phase (up to years) and the symmetrical joint involvement often observed in human RA [31]. Other commonly used animal models include adjuvant-induced arthritis (AIA) and streptococcal cell wall arthritis models, but the CIA model has features that better resemble human RA. For instance, arthritogenic antibodies and strong B-cell responses are observed in RA, and both contribute to the CIA disease progression. However, these are not present in the AIA model [32]. Our previous publication has shown that the CIA Lewis rat model is preferable to the AIA model for PK/PD assessment of RA therapeutics [15], and therefore this animal model was selected in the current study.
Assessments of biomarkers and disease endpoints in control and drug-treated CIA rats allow the development of mechanistic models, which can describe the natural disease progression as well as the PD effects of drugs. Although the etiology and pathophysiology of human RA is more diverse than that in CIA rats, the mechanistic model can capture the major features of the disease and serve as a simplified yet scientifically-based tool for understanding human RA. The mechanism of action of various therapeutics can also be characterized by the mechanistic model. Such translational modeling practices have been extensively used for studying inflammatory conditions and characterizing the properties of anti-inflammatory therapeutic agents [33].
The PD model used in our study was first developed to describe the effects of etanercept on paw edema in CIA rats [13]. Paw sizes of healthy rats were observed to follow a slow and linear type of growth reflected by the parameter k grow . The disease progression component was based on an indirect response model (IDR), where k in and k out represent the production and loss processes of paw edema. The disease condition was assumed to be active in the system starting at time t onset , after which a rapid raise of paw edema was observed. Paw edema in CIA rats peaked around day 21 after collagen induction and began to decline naturally afterwards. The natural remission was represented by the linear function R deg , as it negatively regulates the production process k in . Since abatacept functions as a co-stimulation blocker that affects T-cell activation and the subsequent cytokine cascade, it is reasonable to assume that the mechanism of action of abatacept is similar to etanercept in inhibiting paw edema production. Thus, the same PD model should be appropriate to describe the effects of abatacept in CIA rats. Application of the IDR type model not only has the advantage of revealing parameters regarding both the capacity (I max ) and sensitivity (IC 50) of drug effects, but also allows the comparison of the efficacy of abatacept with other previously studied therapeutic agents (to be discussed). Our results showed that the model was able to capture the disease progression and PD of abatacept in CIA rats. Estimates of most DIS-related parameters, such as t onset and k grow , were close to the previously reported values. However, the baseline rat paw volume (Paw 0) in our current study (55.4 mm2) was slightly lower than those in the etanercept (~62.6 mm2) [13] and anakinra (57.1 mm2) studies [14]. This reflects batch-to-batch differences in the rats and emphasized the importance of considering inter-individual variability in our modeling analysis.
One caveat in our previous etanercept analysis was that a naïve pooling approach left considerable variability in the data that could not be explained. The current analysis adopts population modeling to improve our understanding of sources of variability. The random effects in the k grow , k out , k 0 in , and Paw 0 parameters were assessed. It is reasonable to observe inter-individual variability in k grow and Paw 0, as individual rats may have different natural growth rates and initial paw sizes. The inter-individual variability terms on k out and k 0 in reflect the different immune responses of rats to collagen. Rats react diversely to the collagen challenge. The success rate of inducing CIA in rats is only about 50 %, and those that become arthritic may exhibit swelling in one leg or in both. The severity of paw edema also differs among the group, and therefore it is plausible for the rats to have considerable differences in k out and k 0 in parameters. Variability was not significant for t onset , which was reasonable as all the paw increases have similar onset times (around day 14).
Abatacept has demonstrated safety and efficacy in treating RA in humans [34–37]. In pre-clinical studies, it was shown to prevent arthritis development and bone destruction in CIA rats when used prophylactically [38]. Efficacy of abatacept in a CIA mouse model was reported as it ameliorated T helper cell 1 (TH1) responses [39]. The drug also elicited immunosuppressive effects in cynomolgus monkeys immunized with sheep red blood cells [21]. All these reports suggested good efficacy of abatacept in treatment of arthritis. However, in our study abatacept was far less efficacious.
In vitro study has shown that a 1.0 μg/mL concentration results in significant inhibition of T cell proliferation, and the maximal effects occur at 10 μg/mL [40]. This finding became the basis of further dosage selection in clinical studies, where the trough concentration (C min ) was regarded as the most important factor in dose determination because maintaining a prolonged efficacious exposure at the site of action is critical for anti-rheumatic drugs [25]. Abatacept dosage in humans was determined based on the relationship between ACR20 (American College of Rheumatology Criteria for 20 % improvement) and C min . The steady-state mean (range) is 24 (1–66) μg/mL after multiple IV infusions and 32.5 (6.6–113.8) μg/mL after SC doses [9, 25]. Our estimated IC 50 value (0.731 μmol/L, circa 67.3 μg/mL) is both within the expected clinically effective concentration range and our dosing regimens produced plasma concentrations exceeding this value (Fig. 2).
In our previous report assessing effects of etanercept in CIA rats we hypothesized that multiple-dosing regimens could improve the treatment outcome [13]. Therefore, such dosing groups were added to our subsequent studies with anakinra [14], MTX [41], and abatacept. The doses selected in our studies were based on previous findings that showed successful use of the drug in similar animal models [39, 42, 43]. In this study, the doses selected were even higher than the previously shown efficacious dose in the CIA model [39]. Nevertheless, our experimental results were not as expected as anakinra, MTX, and abatacept all showed modest effects (I max values of 0.279, 0.160, and 0.161) on reduction of paw edema. Our modeling results indicated that IC 50 concentrations (49.4 ng/mL for anakinra, 0.712 nM for MTX, and 0.731 μmol/L for abatacept) were exceeded with the experimental dosing regimens, so dose selection may not be a reason for lack of efficacy and further increases in dose should not produce stronger effects.
One explanation for the difference in our results compared to others may be the starting time of the treatments. In previous studies that showed significant drug effects [39, 42, 43], dosing was initiated shortly after disease onset (on day 1 of arthritis symptoms). Figure 6 shows model simulations of situations where SC multiple-dosing of abatacept (20 mg/kg on the first day followed by 4 more 10 mg/kg doses every 2 days) were assumed to start on different days post-induction. Simulations were performed to generate profiles of paw edema when SC abatacept is started on days 14, 16, 18, or 21. It can be seen that even though abatacept has modest effects, greater suppression of paw edema may be attained with the earlier dosing schedule. The peak of paw edema decreases to around 110 mm2 when dosing starts at day 14 compared with 120 mm2 in the control group. This is in agreement with previous recommendations of early intervention of biologics in RA treatment for better therapeutic outcomes and overall disease control [44, 45].

Model simulations of paw edema disease progression of CIA rats under vehicle control and multiple SC abatacept dosing starting at different days post-induction
The current dosing schedule that started at day 21 post-induction was selected initially because our interest was in regard to the therapeutic (not preventive) effects of the drug. Our previous study with DEX indicated that paw edema peaks at day 21 and can be completely suppressed by DEX given on day 21 [12]. In this animal model, multiple pro-inflammatory cytokines are responsible for producing paw edema and direct inhibition of these cytokines appears most effective in suppressing symptoms. DEX inhibits gene expression of TNF-α, IL-1β, and IL-6, and thus is more effective than the biologics which block only one of these cytokines. Our study results with etanercept, anakinra, and abatacept also agreed with the previous DEX study that TNF-α inhibitors are the most effective protein therapeutic agent for suppressing paw swelling in this animal model [12], and this is indicated by the greater inhibitory effect of etanercept (I max = 0.289) [13] compared with that of anakinra (I max = 0.279) [14], MTX (I max = 0.160) [41], and abatacept (I max = 0.161).
Another possibility for the good efficacy of DEX may be because DEX, as a lipid-soluble small molecule, has better penetration to peripheral sites of inflammation such as the rat paw. The other evidence supporting this hypothesis came from our study with MTX, which is considered the “gold standard” drug for RA. MTX presumably causes apoptosis of activated lymphocytes and adenosine [46] and requires further metabolism within the red blood cells for exerting its effects [41, 47]. It only partly inhibits production of some cytokines (mainly TNF-α) and is not effective on others [48], which is likely the reason why it showed only modest effects on paw edema in CIA rats [41].
It is important to keep in mind that even though DEX is effective on paw edema reduction, it also has other toxicity issues such as bone loss [49]. Biologics, such as etanercept and other TNF-α blockers [50], may provide greater safety in this aspect. Taking into account the efficacy and toxicity features of different therapeutic agents is crucial for RA treatment and the dose optimization of combinations of multiple agents may be necessary for the best therapeutic outcomes.
Conclusions
The pharmacokinetics and pharmacodynamics of abatacept in CIA rats were well described by the proposed population PK/PD/DIS model. A two-compartment model with linear CL characterized the time-concentration profiles of abatacept for the three dosing groups. For SC dosing groups, the absorption rate was modeled as zero-order input with F = 59.2 %. Effects of abatacept on paw volume in CIA rats are modest, but reasonably characterized by our PK/PD/DIS model. Treatment of RA with abatacept (and possibly other biologics) should begin as early as possible in order to maximize the therapeutic benefits.
References
Klareskog L, Catrina AI, Paget S (2009) Rheumatoid arthritis. Lancet 373(9664):659–672
Gregersen PK, Silver J, Winchester RJ (1987) The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 30(11):1205–1213
Corrigall VM, Panayi GS (2002) Autoantigens and immune pathways in rheumatoid arthritis. Crit Rev Immunol 22(4):281–293
Cromartie WJ, Craddock JG, Schwab JH, Anderle SK, Yang CH (1977) Arthritis in rats after systemic injection of streptococcal cells or cell walls. J Exp Med 146(6):1585–1602
Mueller DL, Jenkins MK, Schwartz RH (1989) Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol 7:445–480
Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA (1991) Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med 173(3):721–730
Gaffo A, Saag KG, Curtis JR (2006) Treatment of rheumatoid arthritis. Am J Health Syst Pharm 63(24):2451–2465
Goldsby RA, Kindt TJ, Osborne BA (2000) Kuby immunology, 4th edn. W.H. Freeman & Company, New York, 553 pp
Bristol-Myers Squibb Inc., (2011) Full prescribing information for ORENCIA (abatacept)
FDA (2006) Drug Approval Package Orencia (Abatacept) Injectable (IV). http://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/125118_s0000_OrenciaTOC.cfm
Earp JC, DuBois DC, Molano DS, Pyszczynski NA, Keller CE, Almon RR, Jusko WJ (2008) Modeling corticosteroid effects in a rat model of rheumatoid arthritis I: mechanistic disease progression model for the time course of collagen-induced arthritis in Lewis rats. J Pharmacol Exp Ther 326(2):532–545
Earp JC, DuBois DC, Molano DS, Pyszczynski NA, Almon RR, Jusko WJ (2008) Modeling corticosteroid effects in a rat model of rheumatoid arthritis II: mechanistic pharmacodynamic model for dexamethasone effects in Lewis rats with collagen-induced arthritis. J Pharmacol Exp Ther 326(2):546–554
Lon HK, Liu D, Zhang Q, DuBois DC, Almon RR, Jusko WJ (2011) Pharmacokinetic-pharmacodynamic disease progression model for effect of etanercept in Lewis rats with collagen-induced arthritis. Pharm Res 28(7):1622–1630
Liu D, Lon HK, DuBois DC, Almon RR, Jusko WJ (2011) Population pharmacokinetic-pharmacodynamic-disease progression model for effects of anakinra in Lewis rats with collagen-induced arthritis. J Pharmacokinet Pharmacodyn 38(6):769–786
Earp JC, Dubois DC, Almon RR, Jusko WJ (2009) Quantitative dynamic models of arthritis progression in the rat. Pharm Res 26(1):196–203
Bailer AJ (1988) Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm 16(3):303–309
Nedelman JR, Gibiansky E, Lau DT (1995) Applying Bailer’s method for AUC confidence intervals to sparse sampling. Pharm Res 12(1):124–128
Beal S, Sheiner LB, Boeckmann A, Bauer RJ (2009) NONMEM User’s Guides. (1989–2009). Icon Development Solutions, Ellicott City, MD, USA
Team RDC (2008) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Srinivas NR, Shyu WC, Weiner RS, Warner G, Comereski C, Tay LK, Greene DS, Barbhaiya RH (1997) Assessment of dose proportionality, absolute bioavailability, and immunogenicity response of CTLA4Ig (BMS-188667), a novel immunosuppressive agent, following subcutaneous and intravenous administration to rats. Pharm Res 14(7):911–916
Srinivas NR, Weiner RS, Warner G, Shyu WC, Davidson T, Fadrowski CG, Tay LK, Lee JS, Greene DS, Barbhaiya RH (1996) Pharmacokinetics and pharmacodynamics of CTLA4lg (BMS-188667), a novel immunosuppressive agent, in monkeys following multiple doses. J Pharm Sci 85(1):1–4
Ma Y, Lin BR, Lin B, Hou S, Qian WZ, Li J, Tan M, Ma J, Li BH, Wang H, Wen AD, Guo YJ (2009) Pharmacokinetics of CTLA4Ig fusion protein in healthy volunteers and patients with rheumatoid arthritis. Acta Pharmacol Sin 30(3):364–371
Srinivas NR, Shyu WC, Weiner RS, Tay LK, Greene DS, Barbhaiya RH (1995) Pharmacokinetics of CTLA4Ig (BMS-188667), a novel immunosuppressive agent, following intravenous and subcutaneous administration to mice. J Pharm Sci 84(12):1488–1489
Srinivas NR, Weiner RS, Shyu WC, Calore JD, Tritschler D, Tay LK, Lee JS, Greene DS, Barbhaiya RH (1996) A pharmacokinetic study of intravenous CTLA4Ig, a novel immunosuppressive agent, in mice. J Pharm Sci 85(3):296–298
Hasegawa M, Imai Y, Hiraoka M, Ito K, Roy A (2011) Model-based determination of abatacept exposure in support of the recommended dose for Japanese rheumatoid arthritis patients. J Pharmacokinet Pharmacodyn 38(6):803–832
Roy A, Mould DR, Wang XF, Tay L, Raymond R, Pfister M (2007) Modeling and simulation of abatacept exposure and interleukin-6 response in support of recommended doses for rheumatoid arthritis. J Clin Pharmacol 47(11):1408–1420
Vugmeyster Y, Xu X, Theil FP, Khawli LA, Leach MW (2012) Pharmacokinetics and toxicology of therapeutic proteins: advances and challenges. World J Biol Chem 3(4):73–92
Genovese MC, Covarrubias A, Leon G, Mysler E, Keiserman M, Valente R, Nash P, Simon-Campos JA, Porawska W, Box J, Legerton C 3rd, Nasonov E, Durez P, Aranda R, Pappu R, Delaet I, Teng J, Alten R (2011) Subcutaneous abatacept versus intravenous abatacept: a phase IIIb noninferiority study in patients with an inadequate response to methotrexate. Arthritis Rheum 63(10):2854–2864
Ismael G, Hegg R, Muehlbauer S, Heinzmann D, Lum B, Kim SB, Pienkowski T, Lichinitser M, Semiglazov V, Melichar B, Jackisch C (2012) Subcutaneous versus intravenous administration of (neo)adjuvant trastuzumab in patients with HER2-positive, clinical stage I-III breast cancer (HannaH study): a phase 3, open-label, multicentre, randomised trial. Lancet Oncol 13(9):869–878
Zhuang Y, Xu Z, Frederick B, de Vries DE, Ford JA, Keen M, Doyle MK, Petty KJ, Davis HM, Zhou H (2012) Golimumab pharmacokinetics after repeated subcutaneous and intravenous administrations in patients with rheumatoid arthritis and the effect of concomitant methotrexate: an open-label, randomized study. Clin Ther 34(1):77–90
Schurgers E, Billiau A, Matthys P (2011) Collagen-induced arthritis as an animal model for rheumatoid arthritis: focus on interferon-gamma. J Interferon Cytokine Res 31(12):917–926
Holmdahl R, Lorentzen JC, Lu S, Olofsson P, Wester L, Holmberg J, Pettersson U (2001) Arthritis induced in rats with nonimmunogenic adjuvants as models for rheumatoid arthritis. Immunol Rev 184:184–202
Lon HK, Liu D, Jusko WJ (2012) Pharmacokinetic/pharmacodynamic modeling in inflammation. Crit Rev Biomed Eng 40(4):295–312
Korhonen R, Moilanen E (2009) Abatacept, a novel CD80/86-CD28 T cell co-stimulation modulator, in the treatment of rheumatoid arthritis. Basic Clin Pharmacol Toxicol 104(4):276–284
Maxwell LJ, Singh JA (2010) Abatacept for rheumatoid arthritis: a cochrane systematic review. J Rheumatol 37(2):234–245
Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, Williams GR, Becker JC, Hagerty DT, Moreland LW (2003) Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med 349(20):1907–1915
Kremer JM, Dougados M, Emery P, Durez P, Sibilia J, Shergy W, Steinfeld S, Tindall E, Becker JC, Li T, Nuamah IF, Aranda R, Moreland LW (2005) Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum 52(8):2263–2271
Kliwinski C, Kukral D, Postelnek J, Krishnan B, Killar L, Lewin A, Nadler S, Townsend R (2005) Prophylactic administration of abatacept prevents disease and bone destruction in a rat model of collagen-induced arthritis. J Autoimmun 25(3):165–171
Webb LM, Walmsley MJ, Feldmann M (1996) Prevention and amelioration of collagen-induced arthritis by blockade of the CD28 co-stimulatory pathway: requirement for both B7-1 and B7-2. Eur J Immunol 26(10):2320–2328
Davis PM, Nadler SG, Stetsko DK, Suchard SJ (2008) Abatacept modulates human dendritic cell-stimulated T-cell proliferation and effector function independent of IDO induction. Clin Immunol 126(1):38–47
Liu DY, Lon HK, Wang YL, DuBois DC, Almon RR, Jusko WJ (2013) Pharmacokinetics, pharmacodynamics and toxicities of methotrexate in healthy and collagen-induced arthritic rats. Biopharm Drug Dispos 34(4):203–214
Bendele A, McAbee T, Sennello G, Frazier J, Chlipala E, McCabe D (1999) Efficacy of sustained blood levels of interleukin-1 receptor antagonist in animal models of arthritis: comparison of efficacy in animal models with human clinical data. Arthritis Rheum 42(3):498–506
Schett G, Middleton S, Bolon B, Stolina M, Brown H, Zhu L, Pretorius J, Zack DJ, Kostenuik P, Feige U (2005) Additive bone-protective effects of anabolic treatment when used in conjunction with RANKL and tumor necrosis factor inhibition in two rat arthritis models. Arthritis Rheum 52(5):1604–1611
Castro-Rueda H, Kavanaugh A (2008) Biologic therapy for early rheumatoid arthritis: the latest evidence. Curr Opin Rheumatol 20(3):314–319
Salt E, Crofford L (2012) Rheumatoid arthritis: new treatments, better outcomes. Nurse Pract 37(11):16–22 (quiz 23)
Colmegna I, Ohata BR, Menard HA (2012) Current understanding of rheumatoid arthritis therapy. Clin Pharmacol Ther 91(4):607–620
Chan ES, Cronstein BN (2010) Methotrexate–how does it really work? Nat Rev Rheumatol 6(3):175–178
Neurath MF, Hildner K, Becker C, Schlaak JF, Barbulescu K, Germann T, Schmitt E, Schirmacher P, Haralambous S, Pasparakis M, Meyer Zum Buschenfelde KH, Kollias G, Marker-Hermann E (1999) Methotrexate specifically modulates cytokine production by T cells and macrophages in murine collagen-induced arthritis (CIA): a mechanism for methotrexate-mediated immunosuppression. Clin Exp Immunol 115(1):42–55
Summey BT, Yosipovitch G (2006) Glucocorticoid-induced bone loss in dermatologic patients: an update. Arch Dermatol 142(1):82–90
Seriolo B, Paolino S, Sulli A, Ferretti V, Cutolo M (2006) Bone metabolism changes during anti-TNF-alpha therapy in patients with active rheumatoid arthritis. Ann N Y Acad Sci 1069:420–427
Acknowledgments
This work was supported by funding from the UB Center for Protein Therapeutics, NIH Grant GM 24211, a fellowship for Ms. Lon from Amgen, Inc., and fellowship support for Dr. Liu from Hoffman-La Roche Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lon, HK., Liu, D., DuBois, D.C. et al. Modeling pharmacokinetics/pharmacodynamics of abatacept and disease progression in collagen-induced arthritic rats: a population approach. J Pharmacokinet Pharmacodyn 40, 701–712 (2013). https://doi.org/10.1007/s10928-013-9341-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-013-9341-1