
Abstract
This study investigates the impact of disease on nicotinic acid (NiAc)-induced changes in plasma concentrations of non-esterified fatty acids (NEFA). NiAc was given by constant intravenous infusion to normal Sprague–Dawley and obese Zucker rats, and arterial blood samples were taken for analysis of NiAc, NEFA, insulin and glucose plasma concentrations. The intravenous route was intentionally selected to avoid confounding processes, such as absorption, following extravascular administration. Data were analyzed using nonlinear mixed effects modeling (NONMEM, version VI). The disposition of NiAc in the normal rats was described by a two-compartment model with endogenous synthesis of NiAc and two parallel capacity-limited elimination processes. In the obese rats disposition was described by a one-compartment model with endogenous synthesis of NiAc and one capacity-limited elimination process. The plasma concentration of NiAc drove NEFA (R) turnover via an inhibitory drug-mechanism function acting on the formation of NEFA. NEFA turnover was described by a feedback model with a moderator distributed over a series of transit compartments, where the first compartment (M 1 ) inhibited the formation of R and the last compartment (M N ) stimulated the loss of R. All processes regulating plasma NEFA concentrations were assumed to be captured by the moderator function. Differences in the pharmacodynamic response of the two strains included, in the obese animals, an increased NEFA baseline, diminished rebound and post-rebound oscillation, and a more pronounced slowly developing tolerance during the period of constant drug exposure. The feedback model captured the NiAc-induced changes in NEFA response in both the normal and obese rats. Differences in the parameter estimates between the obese and normal rats included, in the former group, increases in R 0 , k in and p by 44, 41 and 78 %, respectively, and decreases in k out and γ by 64 and 84 %, respectively. The estimates of k tol and IC 50 were similar in both groups. The NiAc–NEFA concentration–response relationship at equilibrium was substantially different in the two groups, being shifted upwards and to the right, and being shallower in the obese rats. The extent of such shifts is important, as they demonstrate the impact of disease at equilibrium and, if ignored, will lead to erroneous dose predictions and, in consequence, poorly designed studies. The proposed models are primarily aimed at screening and selecting candidates with the highest potential of becoming a viable drug in man.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dyslipidemia is present when the concentration of one or more plasma lipoproteins is abnormal. Common lipid abnormalities include elevated levels of total cholesterol, low-density lipoprotein (LDL), cholesterol and triglycerides (TG), and low levels of high-density lipoprotein (HDL) cholesterol. These abnormalities can be found alone or in combination [1] and are a major risk factor for coronary heart disease and other forms of atherosclerotic vascular disease [2]. As a treatment for dyslipidemia in humans, oral doses of 1–3 grams of nicotinic acid (NiAc) per day lower total cholesterol, LDL cholesterol, very-low-density lipoprotein cholesterol and TG, and simultaneously raise HDL cholesterol [3–10]. These beneficial effects were long attributed to the free fatty acid hypothesis, i.e. the ability of NiAc to inhibit the hydrolysis of TG to non-esterified fatty acids (NEFA) and glycerol in adipose tissue, and consequently to reduce plasma NEFA concentrations [11]. This enduring theory was recently challenged, suggesting that the prominent NiAc-induced lipoprotein modification is independent of both GPR109A and NEFA suppression [12]. However, it has been shown that inhibition of lipolysis via GPR109A by NiAc or its analogues acutely improve glucose use and insulin sensitivity in subjects with type 2 diabetes mellitus [13–17]. Various endogenous regulators, including insulin, glucose, glucagon, growth hormone, cortisol and TG, are known to be involved in NEFA homeostasis [18–20]. Insulin, the main hormonal regulator of NEFA, rapidly inhibits the hydrolysis of TG to NEFA and glycerol [21, 22], and stimulates the re-esterification of NEFA to TG [22, 23] via a slower process.
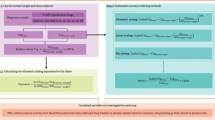
Recently, we published a feedback model describing NiAc-induced changes in plasma NEFA concentrations in normal Sprague–Dawley rats [24]. Subsequently, we evaluated this model quantitatively [25], and challenged it by extending the data set with intravenous and oral dosing regimens that resulted in different NiAc exposure patterns [26]. Our feedback model was based on mechanistic principles that mimicked insulin’s regulatory impact on NEFA homeostasis by introducing a moderator acting via a series of transit compartments, with the moderator in the first and last compartments representing the rapid inhibitory and slow stimulatory regulation mechanisms, respectively (Fig. 1). Because underlying disease contributes to the large interindividual variability in a drug’s pharmacodynamics [27], we have now used the obese Zucker rat (fa/fa) [28], an animal model of insulin resistance and metabolic disorders, to determine if disease affects the pharmacokinetics or pharmacodynamics, and to further challenge the feedback model. Our previous data from normal Sprague–Dawley rats [26] have been used as reference.

Schematic illustration of the disposition of NiAc and the feedback model describing NiAc-induced changes in NEFA in obese Zucker rats. a Disposition of NiAc. C p denotes the NiAc concentration in plasma and Inf the intravenous infusion. The NiAc disposition parameters are defined in Table 1. b The feedback model describing NiAc-induced changes in plasma NEFA. NEFA and M 1 ,…,M N denote the response and moderator compartments, respectively. The NEFA turnover parameters are defined in Table 2. I(C p ) is defined in Eq. (2 ). The number of moderator transit compartments N was 8. The solid and dashed lines represent fluxes and control processes, respectively
Obesity is frequently associated with insulin resistance and known to influence the distribution and clearance of compounds [29–33]. Furthermore, insulin is involved in the regulation of NEFA turnover. Thus, it is possible that NEFA homeostasis may be altered in patients suffering from insulin resistance and that disease-induced hormonal changes may influence the onset, intensity and duration of drug effects by altering NEFA turnover. Therefore, during the model building process it is essential to assess the impact of disease on the structural pharmacokinetic-pharmacodynamic (PKPD) model, and on the system parameters, drug parameters and pharmacokinetics. However, in the clinic it is often difficult to ascertain whether a disease-associated change in the response profile of a drug has a purely pharmacokinetic or pharmacodynamic basis. This difficulty is intensified by the unstable nature of most diseases and by the fact that few patients suffer from only one well defined disease. Therefore, studies of the kinetics of drug action in disease states in animal models under well defined conditions are crucial [34].
Whereas lean Zucker rats have lipoprotein and lipid patterns similar to the Sprague–Dawley strain, the obese Zucker rat is characterized by mild hyperglycemia [35, 36] and a marked increase in plasma lipoproteins and lipids, including NEFA [28]. Obese Zucker rats also have pronounced hormonal changes, with increased plasma insulin levels [37–39] and insulin-resistance [40], plus impaired metabolism of glucagon [41] and growth hormone [38, 39, 42].
In this study, NiAc-induced changes in plasma NEFA concentrations were explored after different rates and durations of intravenous NiAc infusion. The exposure–response relationships were determined and quantified on the basis of integrated PKPD modeling, and used together with the parameter estimates to detect differences between the normal and disease-model animals.
Materials and methods
Chemicals
Nicotinic acid (pyridine-3-carboxylic acid) was obtained from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in saline (0.9 % NaCl). All solvents were of analytical grade and the water used in the experiments was obtained from a water purification system (Elgastat Maxima, ELGA, Lane End, UK).
Animals and surgical procedures
Male obese Zucker (fa/fa) and normal male Sprague–Dawley rats were purchased from Harlan Laboratories B.V. (The Netherlands) at 7 and 11 weeks of age, respectively, and used at 16 weeks of age. The animals were housed in groups of 5–6 with free access to standard rodent chow (R3, Laktamin AB, Stockholm, Sweden) and tap water. They were kept in climate-controlled facilities at room temperature 20–22 °C and relative humidity 40–60 % under a 12:12-h light:dark cycle (lights on 6:00 am). The study was approved by the Ethics Committee for Animal Experiments, Göteborg, Sweden.
Surgery was performed under isoflurane (Forene®, Abbott Scandinavia AB, Sweden) anesthesia and the body temperature maintained at 37 °C using a thermoregulated heating pad. Catheters were implanted in the left carotid artery for blood sampling and right external jugular vein for drug administration, according to Popovic and Popovic [43]. Prior to cannulation, catheters (Intramedic®, PE50, Becton–Dickinson and Company, USA) were filled with sterile sodium-citrate solution (20.6 mM Na3-citrate in sterile saline; Pharmaceutical and Analytical R&D, AstraZeneca, Mölndal, Sweden) to prevent clotting. After cannulation, the catheters were exteriorized at the nape of the neck and sealed. After surgery, rats were housed individually and allowed a minimum of 5 days recovery before initiation of the experiments.
Experimental design
Animals were deprived of food for 14 h prior to dosing and throughout the experiment to minimize the fluctuations in NEFA caused by food intake, but they had free access to drinking water. On the day of experimentation, each animal was weighed and moved to a clean cage, and its venous catheter was connected to an infusion pump (CMA 100, Carnegie Medicin AB, Stockholm, Sweden). Following a 30 min adaptation period, two consecutive blood samples were collected 15 and 5 min prior to drug administration to enable determination of predose baseline NEFA, NiAc, insulin and glucose concentrations.
Rats (Sprague–Dawley weighing 220–367 g, obese Zucker weighing 473–547 g on the day of experimentation) were randomly allocated to parallel groups and given an intravenous infusion of 20 μmol kg−1 NiAc (n = 9 Sprague–Dawley, n = 8 Zucker) or vehicle (0.9 % NaCl, n = 10 Sprague–Dawley, n = 2 Zucker) over 30 min, or of 51 μmol kg−1 NiAc (n = 7 both strains) or vehicle (n = 8 Sprague–Dawley, n = 2 Zucker) over 300 min. Dosing solutions were prepared within 30 min of administration, and control rats received the same infusion volume as rats in the respective NiAc-treated group. Multiple arterial blood samples (11–15 per rat) of 100–120 μL were drawn over 100 min in the 30 min infusion groups, and over 500 min in the 300 min infusion groups for analysis of NiAc and NEFA plasma concentrations. Each sample was replaced with an equal volume of sterile sodium-citrate solution to maintain a constant circulatory volume. The total volume of blood removed per animal was less than 1.5 mL, and sampling procedures were the same in all animals. Blood samples were collected in EDTA coated polyethylene tubes and kept on ice until centrifuged (10,000×g, 1 min, 4 °C). Plasma was stored at −20 °C pending analysis. The onset of infusion was taken as time zero (0 min).
Analytical assays
NiAc in plasma was analyzed and quantified using LC–MS. The high performance liquid chromatography (HPLC) system was an Agilent 1100 Series (Hewlett-Packard GmbH, Walbronn, Germany) coupled to an HTC PAL auto sampler (CTC Analytics AG, Zwingen, Germany). Plasma samples (50 μL per sample) were precipitated with cold acetonitrile containing 0.2 % formic acid (150 μL per sample). After vortex mixing and centrifugation at 4 °C (4,000×g, 20 min), an aliquot of 100 μL of the supernatant was used for the analysis. The mobile phase consisted of (A) 2 % acetonitrile and 0.2 % formic acid in water, and (B) 0.2 % formic acid in acetonitrile. Separation was performed on a 50 × 2.1 mm Biobasic AX column with 5 μm particles (Thermo Hypersil-Keystone, Runcorn, Cheshire, UK) with a gradient of 95–20 % B over 1 min, held at 20 % B for 1.5 min, and returned to initial conditions in one step. The HPLC system was connected to a Sciex API 4000 quadrupole mass spectrometer with a positive electrospray ionization interface (Applied Biosystems, Ontario, Canada) and the mass transition was 124.0 > 80.2. Data acquisition and evaluation were performed using Analyst 1.4.1 (Applied Biosystems). The method showed linearity over a concentration range of 0.001–28 μmol L−1. The lower limit of quantification (LLOQ) was 1 nmol L−1 applying a sample volume of 50 μL plasma.
Plasma NEFA was analyzed using an enzymatic colorimetric method (Wako Chemicals GmbH, Neuss, Germany) adapted to a 96-well format [44]. The method showed linearity over a NEFA concentration range of 0.002–2 mmol L−1, with a LLOQ of 0.002 mmol L−1 applying a sample volume of 10 μL plasma.
Insulin concentrations were determined using a radioimmuno assay (Rat insulin RIA kit, Cat no. RI-13K, Millipore, USA). Glucose concentrations were measured using an enzymatic colorimetric method (Glucose HK CP, kit No: A11A01667, ABX Pentra, France). The glucose assay was performed on a Cobas Mira Analyser (Hoffman-La Roche & Co., Basel, Switzerland).
The binding to plasma proteins was measured by an automated equilibrium dialysis assay run in duplicate. After overnight dialysis of plasma against a phosphate buffer at pH 7.0, plasma and buffer samples were analyzed using LC–MS. Binding was measured at NiAc concentrations of 1 and 10 μmol L−1. The fraction unbound, f u , was calculated from the analysis results of plasma and buffer.
Pharmacokinetic and pharmacodynamic model
The disposition of NiAc in obese Zucker rats was modeled as a one-compartment model with endogenous synthesis (Synt) of NiAc and capacity-limited elimination (Fig. 1), mathematically described as:
where C p denotes the NiAc concentration in the central compartment, V c the central volume of distribution, Input the rate of intravenous infusion of NiAc, and Synt the endogenous synthesis rate. The V max and K m parameters denote the maximal velocity and Michaelis–Menten constant, respectively.
The hydrolysis of TG to NEFA and glycerol in adipocytes is inhibited by NiAc, with this inhibitory process I(C p ) being described by:
where C p , I max , IC 50 and γ are, respectively, the NiAc concentration in plasma, the maximum drug-induced inhibitory effect of NEFA, the NiAc plasma concentration at 50 % reduction (potency) of the NEFA turnover rate, and the sigmoidicity factor.
The feedback is governed by a moderator M which is distributed over a series of eight transit compartments (Fig. 1), where moderator M 1 in the first compartment inhibits the adipocyte-dependent formation of R, and moderator M 8 in the eighth compartment stimulates the loss of R. The dual action of insulin on NEFA regulation is captured firstly by M 1 , which denotes the rapid inhibition of the hydrolysis of TG to NEFA and glycerol in adipocytes [21, 22], and secondly by M 8 , which represents the delayed stimulation of re-esterification of NEFA to TG [22, 23]. The moderator is affected by R via a first-order build-up of M (k tol ·R). Each transit compartments has a transit time of 1/k tol .
When NiAc inhibits the adipocyte-dependent formation of NEFA, NEFA will decrease, causing a reduction in the production of moderator and a subsequent decrease in M 1 . As the formation of NEFA is inversely proportional to the moderator raised to the power of p (M p 1 ), the formation of NEFA will increase when M 1 decreases. After a delay, the level of moderator M 8 in the final compartment will also fall, reducing the loss of NEFA. Eventually the concentrations of R and M i (where i = 1,…, 8) will equilibrate.
Due to lipoprotein lipase-catalyzed hydrolysis of TG to NEFA and glycerol in the capillaries, a lower physiological limit of NEFA was observed at high NiAc concentrations in normal rats [26]. This process is initially incorporated as a zero-order production term k cap in the modeling of the obese rats:
where M 1 and M 8 are described above, k in is the turnover rate, p the amplification factor, I(C p ) the inhibitory drug mechanism function, k cap the formation of NEFA in capillaries, and k out the fractional turnover rate of R. The turnover of the moderators is given by:
The turnover rate k in of NEFA is expressed as:
where R 0 is the baseline response.
The steady state values for response R and moderators M i (i = 1,…, N) are given by:
where R ss is the unique solution of the equation:
in which k in is defined in Eq. (5) and R SS is the steady state response. In general it is not possible to write the solution R SS of Eq. (7) as an explicit expression when k cap is nonzero.
Initial parameter estimates and regression model selection
The initial estimates of R 0 , and k out for the obese Zucker rats were derived graphically. The baseline concentration R 0 was based on the predose level of NEFA and R 0 could be approximated to around 1.1 mmol L−1. Following a high dose of NiAc (C p ≫ IC 50 ), Eq. (3) is approximated by:
Initially k cap was assumed to be much less than \( k_{out} \cdot R \cdot R_{0} \), so Eq. (8) can be simplified to:
where M 8 is approximated by R 0 . Thus, the initial downswing of R on a semi-logarithmic plot gives a slope of −k out ·R 0 (k out ≈ 0.05 L mmol−1 min−1).
As a lower physiological limit of NEFA was not seen in the experimental data of the obese Zucker rats, the k cap parameter may be difficult to estimate during disease. However, the estimate of k cap from the analysis of the normal animals was used as an initial estimate. The initial estimates of k tol and IC 50 were also approximated according to the final estimates in normal rats. The sigmoidicity (γ) and amplification (p) factors were initially both set equal to unity. As in our previous studies [24, 26], the number of transit compartments N was eight.
NiAc and NEFA time-courses were modeled using the nonlinear mixed effects approach as implemented in nonlinear mixed effects modeling (NONMEM) version VI level 2.1 (Icon Development Solutions, Maryland, USA). The Laplacian estimation method was used throughout the pharmacokinetic and pharmacodynamic model building processes. For all pharmacokinetic and pharmaco-dynamic parameters, interindividual variability was modeled as exponential models and random residual variability as proportional models. The individual pharmacokinetic parameters were introduced as fixed parameters in the combined PKPD analysis. Model selection was based on descriptiveness of the experimental data, the objective function value (OFV) and the precision of parameter estimates, and it was explored visually in diagnostic plots using Census, version 1.1 (Novartis Pharma AG, Basel Switzerland).
Results
Physiological characteristics
As shown in Table 1, the physiological characteristics were similar in our normal Sprague–Dawley rats and in the lean Zucker rats used by van Schaick et al. [45], but differed markedly in the obese Zucker rats. Total body weight and NEFA concentrations were both ~70 % higher in the obese rats than in normal rats, and the obese rats were hyperinsulinemic, even though their glucose concentrations were similar to those in normal rats. Although the endogenous mean NiAc concentration was higher in obese rats, it was below the LLOQ in some obese animals. Their mean baseline concentration is therefore based on a limited number of samples, and as a result it is uncertain and probably overstated.
NiAc disposition
Observed NiAc concentration time-courses for the obese and normal rats are depicted in Fig. 2. NiAc infusion rates of 0.67 μmol min−1 kg−1 (20 μmol kg−1 over 30 min) and 0.17 μmol min−1 kg−1 (51 μmol kg−1 over 300 min) resulted in slightly higher plasma concentrations in the obese rats than in the normal rats. The highest individual NiAc concentrations were 23.3 and 17.9 μmol L−1 in obese and normal rats, respectively.

Observed plasma NiAc concentration–time profiles in normal (dotted lines) and obese (solid lines) rats during and after vehicle (grey) or NiAc (black) administration. Control groups received vehicle according to the same regimen as their respective NiAc-treated group. a 30 min infusion of 0 or 20 μmol kg−1 NiAc per kg of body weight; b 300 min infusion of 0 or 51 μmol kg−1 NiAc. The NiAc baseline concentrations could only be quantified in a limited number of control samples as they were below the LLOQ (<0.001 μmol L−1)
The disposition parameters of NiAc in normal [26] and obese rats are summarized in Table 2.
The aim of modeling NiAc concentration–time data was to obtain individually fitted time-courses that could drive the NEFA concentration–time data. The disposition of NiAc in obese rats was adequately described by a one-compartment model with endogenous NiAc synthesis (Fig. 3). The diagnostic plots of the obese rat data in Fig. 4 confirmed the consistency between observed and predicted NiAc concentrations, and there was no marked trend in the residual plots. The disposition parameters were estimated with high precision, but the interindividual variability was poorly estimated in this preliminary analysis.

Four representative model fits of NiAc plasma concentration–time data in obese Zucker rats after a 30 min infusion of 20 μmol kg−1 NiAc or 300 min infusion of 51 μmol kg−1 NiAc. Solid and dotted lines represent individual and population fits, respectively

Goodness-of-fit plots for NiAc in obese Zucker rats. Observed versus population (a) and individually (b) fitted concentration on a logarithmic scale. Individually weighted residuals versus time (c), and individually fitted concentrations on a semi-logarithmic scale (d). Conditional weighted residuals versus time (e), and population fitted concentrations on a semi-logarithmic scale (f)
In both the normal and obese rats, protein binding of NiAc was similar at both 1 and 10 μmol L−1 NiAc. The free fraction in plasma (f u ) at both concentrations was therefore averaged, being 78 % for the normal and 98 % for the obese rats (Table 2).
NiAc-induced changes in NEFA response
The observed individual NEFA concentration–time profiles after NiAc administration are shown for obese and normal rats in Fig. 5, and the observed differences are summarized qualitatively in Table 3. In the normal rats, plasma NEFA concentrations decreased rapidly to a lower physiological limit of 0.055 mmol L−1 [26]. In the obese rats, however, the NEFA reduction was slower and no plateau indicative of a lower physiological limit was reached. The lowest measured NEFA concentration in obese rats was 0.16 mmol L−1. Following cessation of NiAc infusion, the NEFA concentration returned towards baseline in all animals. After the 30 min infusion, it rebounded above baseline in the normal rats but not in the obese rats. In the 300 min infusion groups there was a slowly developing tolerance which was more pronounced in the obese rats than in the normal rats, and after cessation of infusion the NEFA concentration in obese animals returned to baseline more slowly and showed less pronounced rebound and post-rebound oscillation than in normal animals.

Observed plasma NEFA concentration–time profiles in normal (dotted lines) and obese (solid lines) rats during and after vehicle (grey) or NiAc (black) administration. Control groups received vehicle according to the same regimen as their respective NiAc-treated group. a 30 min infusion of 0 or 20 μmol kg−1 NiAc per kg of body weight; b 300 min infusion of 0 or 51 μmol kg−1 NiAc. The NEFA concentrations were stable in individual control animals, but there was large variability between animals
Figure 6 shows representative individual and population predictions of changes in NEFA concentration in obese rats superimposed on experimental data, after different doses and durations of infusion of NiAc. Model predicted and experimental data were consistent, and the model captured the elevated NEFA baseline concentrations, the slowly developing tolerance, the lack of rebound following 30 min infusions, and the small oscillatory rebound following 300 min infusions in the obese rats. Because a lower physiological limit was not reached, k cap could not be estimated with satisfactory precision, so it was fixed to zero throughout the analysis. The diagnostic plots for the data from the obese animals confirmed the consistency of experimental and predicted NEFA concentrations, and there were no marked trends in the residual plots (Fig. 7). The final population parameter estimates and interindividual variability are shown in Table 4.

Four representative model fits of NEFA plasma concentration–time data in obese Zucker rats after 30 min infusion of 20 μmol kg−1 NiAc or 300 min infusion of 51 μmol kg−1 NiAc. Solid and dotted lines represent individual and population fits, respectively

Goodness-of-fit plots for NEFA in obese Zucker rats. Observed versus population (a) and individually (b) fitted concentrations on a logarithmic scale. Individually weighted residuals versus time (c) and individually fitted concentrations (d). Conditional weighted residuals versus time (e) and population fitted concentrations (f)
All primary parameters were estimated with high precision with the exception of IC 50 . The poor precision in IC 50 may reflect the large number of parameters and the limited number of animals.
The equilibrium NiAc concentration–NEFA response relationships of the normal and obese rats were simulated using the final parameter estimates (Table 4) and either Eq. (11) (numerical solution, for normal rat data) or (13) (“Steady state response” in Appendix section, for obese rat data). It can be seen from Fig. 8 that the steady state NEFA concentration R SS for the obese rats was higher than that for the normal rats over the entire range of simulated concentrations. There was a sigmoid relationship between the simulated steady state concentration of NiAc and NEFA in normal animals, with NEFA decreasing as the concentration of NiAc increased from ~0.02 to 0.5 μmol L−1. In obese animals, the NEFA concentration decreased progressively with increasing NiAc concentration, with no evident plateau. In other words, the curve for the obese animals was shifted upwards and to the right, and was shallower, than that for the normal animals.

Simulated steady state plasma NiAc concentration versus predicted plasma NEFA concentration at equilibrium (R SS ) for obese (solid line) and normal (dotted line) rats. These two relationships were derived using Eq. (11) (normal rats) and (13) (obese rats) in “Steady state response” in Appendix section, and the final parameter estimates in Table 4. The plots highlight the difference between the obese and normal rats at low (a), intermediate (around IC 50 (b)) and high (c) NiAc concentrations
The distinguishing characteristics of NEFA dynamics in normal and obese rats are given in a simulation based on mean population parameters in Fig. 9.

Simulated plasma NEFA concentrations following a 300 min constant rate infusion of NiAc (51 μmol kg−1) in normal and obese rats. Data are obtained from the mean population estimates, given in Table 4
Discussion
As obesity causes physiological changes that may influence drug disposition [29–33], and insulin is involved in regulating normal NEFA turnover, it is possible that the NiAc-induced changes in NEFA response is altered in these patients. The purpose of this study was to investigate possible differences in NiAc disposition and NiAc-induced changes in NEFA between normal Sprague–Dawley and obese Zucker rats. The obese rats served as an animal model of insulin resistance and metabolic disorders [46]. The intravenous route was intentionally selected to avoid confounding processes, such as absorption, following extravascular administration.
Physiological characteristics
Because normal Sprague–Dawley rats provide one of the primary screening models in drug discovery, and decisions often are based on such data, it is important to determine how the response to a drug in normal rats translates to that in an animal model of disease and, eventually, in patients. The translational value/potential of non-human disease models still has to be validated for the specific chemical series in patients. Before concluding that any difference in the outcome of NiAc administration is due exclusively to the impact of disease, it would be necessary to eliminate variability originating from differences in the rat strain. However, it has previously been reported that the lipid and lipoprotein profiles in Sprague–Dawley rats and lean Zucker rats are similar [47, 48]. Furthermore, the physiological characteristics of the Sprague–Dawley rats in this study and of the lean Zucker rats in that of van Schaick et al. [45] are in the same range. Therefore we feel confident that the differences we observed between normal Sprague–Dawley and obese Zucker rats following administration of NiAc are a consequence of disease, not of the strain of rat. Although there was a small weight difference between the Sprague–Dawley (present study) and lean Zucker [45] rats, this may reflect their age, 16 and 17 weeks in the two studies, respectively. Furthermore, the higher insulin and lower glucose concentration in the lean Zucker rats compared to the Sprague–Dawley rats may be due to differences such as the length of the fast, age or stress.
The physiological characteristics of the obese Zucker rats differed considerably from both the normal Sprague–Dawley and the lean Zucker rats (Table 1), with the most marked difference being the higher insulin concentration in the obese rats compared to the normal rats (2.9 and 0.017 nmol L−1, respectively). In spite of this, the glucose concentrations were similar in these two groups, which is a clear indication of insulin resistance in the obese animals. The NEFA baseline concentration was ~70 % higher in the obese rats than in the normal Sprague–Dawley rats.
Disposition of NiAc
The disposition of NiAc differed in the obese and normal rats. In normal rats it was described by a two-compartment model with endogenous NiAc synthesis and two parallel capacity-limited elimination pathways [26], whereas in obese rats it was adequately described by a one-compartment model with endogenous NiAc synthesis and a single capacity-limited elimination process. Not only has it been reported that the pharmacokinetics of drugs may be altered in disease [49–51], but it has been proposed that obesity influences the distribution and clearance of compounds [29–33]. The altered disposition of NiAc in obese rats compared to normal rats is, therefore, not surprising. However, the intercompartmental distribution Cl d in normal rats was estimated to 0.00085 L min−1 kg−1 which corresponds to a blood flow in humans around 20 mL min−1. The peripheral compartment would then represent a tissue that is only perfused to a tenth of that of adipose tissue. This indicates that a one-compartment model might suffice to describe the disposition of NiAc in normal rats.
Plasma protein binding of NiAc was slightly lower in the obese rats than in normal rats, which is consistent with the possible competition of NEFA for binding to plasma proteins [52]. Because NiAc is rapidly eliminated and perfusion of the liver is the rate-limiting step, the unbound NiAc concentration is dependent on f u [53], suggesting that a difference in protein binding between normal and obese rats might be important. However, as the molar concentration of plasma albumin is around 600 μmol L−1 and our highest observed NiAc concentration was only around 20 μmol L−1, the impact of protein binding would be negligible and unlikely to account for other differences in the NiAc–NEFA system of the two strains of rat. Although poorly estimated interindividual variability, we achieved our aim in modeling NiAc-concentration time data to find individual time profiles that drove the individual NEFA concentration time-courses.
NiAc-induced changes in NEFA response
We expected that metabolic differences between normal and obese rats would alter the antilipolytic effects of NiAc treatment, as evidenced by changes in the plasma NEFA concentration–time profile. This we found, in spite of the fact that the rats were treated identically (Tables 3, 4; Fig. 5). Infusion of NiAc to normal rats effectively decreased the plasma NEFA concentration to around 10 % of the predose baseline level (see also [26]), and because increasing the infusion rate did not further decrease NEFA concentrations, there seems to be a lower limit of NEFA concentrations in normal rats of around 0.05 μmol L−1. In obese rats, infusion of NiAc also decreased plasma NEFA concentrations. However, when data from equivalent infusion paradigms were compared it was found that the onset was slower in obese than in normal rats, as evidenced by a shallower initial slope in the NEFA concentration–time data, and that the plasma NEFA concentration was only reduced to around 30 % of the predose baseline level. There was also no lower physiological limit, seen as a plateau in normal rats, in the obese rats, presumably because the infusion rate of NiAc was too low. After stopping NiAc infusion, plasma NEFA concentrations increased to 18–260 % above baseline in normal rats, with the degree of rebound appearing to be dependent on both the extent and duration of NiAc exposure [25, 26]. In the obese rats, although NEFA concentrations increased post-infusion, they did not exceed the baseline concentration after a 30 min infusion. After a 300 min infusion, rebound and post-rebound oscillations were observed, but they were substantially less than in the normal rats, and there was more pronounced slowly developing tolerance during the period of constant NiAc exposure in obese than normal rats, resembling the pattern during 300 min infusions in normal rats at lower NiAc doses (5 or 10 μmol kg−1) [24, 26].
Our feedback model was originally developed [24] and challenged [26] to describe NiAc-induced changes in NEFA concentrations in normal rats. It was built upon mechanistic principles, and successfully mimicked insulin’s dual regulatory impact on NEFA homeostasis by incorporating a moderator that acted via a series of transit compartments, with the moderator in the first and last compartment representing the rapid and slow regulation, respectively. Although the pharmacodynamic characteristics of the obese rats in our present study differed from those of normal rats, the feedback model successfully described the pattern of NiAc-induced changes in NEFA in obese rats, capturing the increased NEFA baseline, the pronounced slowly developing tolerance, the diminished rebound, and, following the 300 min infusions, the post-rebound oscillations. However, as the dose was too low, no lower physiological limit of NEFA was achieved, so k cap could not be estimated with acceptable precision in the modeling process. When k cap was fixed to the value of the normal rats, the precision in the rest of the parameters was impaired and the OFV increased, so it was fixed to zero throughout the analysis.
In the obese rats, parameters were generally estimated with high precision and there were no correlations in the residual plots. Compared to the normal rats, R 0 , k in (estimated as a secondary parameter in the analysis) and p were increased by 44, 41 and 78 %, respectively, in the obese rats, and k out andγ were reduced by 64 and 84 %. The k tol and IC 50 were similar in both rat strains.
Insulin inhibits the formation of NEFA (k in , [21, 22]) and stimulates its loss (k out , [22, 23]). Because the NEFA baseline concentration R 0 , is governed by the ratio of k in to k out in obese rats (see Eq. (15) in “Response at baseline” Appendix section), an increase in k in coupled to a decrease in k out would have a multiplicative effect in raising R 0 . Thus, the reduced influence of insulin on NEFA concentrations in insulin-resistant obese Zucker rats may account for the increased R 0 .
The ratio k tol /k out was estimated as 0.085 and 0.30 mmol L−1 in normal and obese rats, respectively. It has been predicted that, as this ratio increases, the extent of rebound should decrease [25]. This is consistent with the reduced rebound and post-rebound oscillations observed in obese rats in this study (Fig. 5). We also found that the amplification factor p was increased in the obese rats, which, according to our previous predictions, would increase the extent of tolerance [25]. Again, this can be seen in the obese rats during the period of constant exposure (Fig. 5). The increased value of p indirectly (Eq. 7) contributed to a decreased slope of the steady state concentration–response relationship (Fig. 8) [25], although this shallow relationship is primarily due to the decreased value of the sigmoidicity factor γ in the obese animals. Correlation matrix, within respective group, showed insignificant dependence between γ and p.
The concentration–response relationships at equilibrium differed in obese and normal rats (Fig. 8), being shifted upwards and to the right, and being shallower, in the obese animals. The extent of such shifts is important, as they demonstrate the impact of disease at equilibrium and, if ignored, will lead to erroneous dose predictions and, in consequence, poorly designed studies. The steady state response for the obese rats at baseline NiAc concentrations was higher than in the normal rats (see a) in Fig. 8). As already discussed, this reflects the multiplicative effect of the increase in k in and decrease in k out on the NEFA baseline concentration. The increased value of p in the obese rats compared to the normal rats is reflected in the upward shift of the curve according to Eq. (14) (“Response at baseline” in Appendix section). At NiAc concentrations equal to IC 50 (see b) in Fig. 8) the steady state response is governed by the ratio of k in to k out and the amplification factor p (Eq. 16 in “Steady state response at IC 50 ” Appendix section). This steady state response was increased in the obese animals because of the multiplicative effect of the increase in k in and decrease in k out , together with the increase in p. At concentrations above 0.5 μmol L−1 the steady state response of the normal rats is governed by the ratio of k cap to k out (Eq. 17 in “Steady state response at high NiAc concentrations” Appendix section). In the obese rats, the sigmoidicity factor γ was estimated to be 0.35 whereas it was 2.2 in normal rats. The smaller estimate of γ in the obese animals implies a lower sensitivity to a change in plasma NiAc concentrations as compared to normal rats (Eq. 13 in “Steady state response” Appendix section, see c in Fig. 8). The steep (normal rats) and shallow (obese rats) concentration–response relationship in Fig. 8 are then manifested as the NEFA—time profiles shown in Fig. 9. Notice, in obese rats, the initial “overshoot”, more pronounced tolerance development, less rebound and post-rebound oscillations.
Several reports indicate that NiAc or its analogues improve glucose use and insulin sensitivity in type 2 diabetic patients, at least in the short term [13–17]. This is in contrast to reports indicating that the long-term administration of NiAc decreases glucose tolerance in these patients [54, 55]. These opposing acute and chronic NiAc-induced effects on glucose control parallel the disparate effects on NEFA lowering. While acute administration results in a rapid reduction in NEFA [13–17], chronic treatment is associated with a NEFA return to pre-treatment levels [54, 55]. A NiAc analogue that, taken prior to a meal, rapidly decreases NEFA concentrations substantially with short effect duration (e.g. around 2 h) and no rebound might be a compound to aim for. With reduced NEFA concentration, glucose will be used as a source of energy, which may result in improved insulin sensitivity [14–16]. Short effect duration has proven to be beneficial for systems exhibiting tolerance, as the primary effect does not last long enough for the counter-acting mechanisms, accountable for tolerance and rebound, to develop [56–61]. Because of the shallow steady state concentration–response relationship seen in the obese rats it may be difficult to find a dose regimen of NiAc with a rapid onset and offset of response and with a substantial decrease of NEFA. However, considering NiAc’s pluripotency [12], future studies to explore whether an optimized NiAc treatment regimen could achieve sustainable NEFA lowering, may indeed alter its therapeutic value and use.
The system parameters in diseased animals may be used to predict the response following administration of NiAc analogues with different drug properties, to find a compound that has the preferred therapeutic profile. Knowledge about how disease alters the effects of a drug facilitates the process of translating drug effects from healthy volunteers to patients, but obviously it would require future studies in healthy and patient populations to evaluate the performance of the model in the clinic.
In conclusion, the pharmacodynamic characteristics of obese and normal rats differed, with obese animals showing an increased NEFA baseline concentration, more pronounced development of tolerance during the period of constant drug exposure, diminished post-infusion rebound, and, following the 300 min infusions, diminished post-rebound oscillation. The feedback model captured the experimental data consistently and showed that the parameters that differed most from the normal animals were R 0 , k in (estimated as a secondary parameter in the analysis), k out , p and γ, with k tol and IC 50 being almost unaltered. The concentration–response relationship at equilibrium was shifted upwards and to the right, and was substantially shallower in the obese animals than it was in normal animals. Information such as this provides a vital aid when predicting drug effects from healthy volunteers to patients. This also emphasizes the importance of using totality of the data across normal rats, appropriate animal disease models and healthy volunteers to project doses in patients.
References
Ballantyne CM, O’Keefe JH, Gotto AM (2009) Dyslipidemia & atherosclerosis essentials, overview of atherosclerosis and dyslipidemia. Jones and Bartlett, Sudbury
National Cholesterol Education Program (2002) Detection, evaluation and treatment of high blood cholesterol in adults. http://www.nhlbi.nih.gov/guidelines/cholesterol/atp3full.pdf
Grundy SM, Mok HYI, Zech L, Berman M (1981) Influence of nicotinic acid on metabolism of cholesterol and triglycerides in man. J Lipid Res 22(1):24–36
Vega GL, Grundy SM (1994) Lipoprotein responses to treatment with lovastatin, gemfibrozil, and nicotinic acid in normolipidemic patients with hypoalphalipoproteinemia. Arch Intern Med 154(1):73–82
Martin-Jadraque R, Tato F, Mostaza JM, Vega GL, Grundy SM (1996) Effectiveness of low-dose crystalline nicotinic acid in men with low high-density lipoprotein cholesterol levels. Arch Intern Med 156(10):1081–1088
Mostaza JM, Schulz I, Vega GL, Grundy SM (1997) Comparison of pravastatin with crystalline nicotinic acid monotherapy in treatment of combined hyperlipidemia. Am J Cardiol 79(9):1298–1301
Guyton JR, Goldberg AC, Kreisberg RA, Sprecher DL, Superko HR, O’Connor CM (1998) Effectiveness of once-nightly dosing of extended-release niacin alone and in combination for hypercholesterolemia. Am J Cardiol 82(6):737–743
Knopp RH, Alagona P, Davidson M, Goldberg AC, Kafonek SD, Kashyap M, Sprecher D, Superko HR, Jenkins S, Marcovina S (1998) Equivalent efficacy of a time-release form of niacin (Niaspan) given once-a-night versus plain niacin in the management of hyperlipidemia. Metab Clin Exp 47(9):1097–1104
Capuzzi DM, Guyton JR, Morgan JM, Goldberg AC, Kreisberg RA, Brusco OA, Brody J (1998) Efficacy and safety of an extended-release niacin (Niaspan): a long-term study. Am J Cardiol 82(12):74–81
Guyton JR, Blazing MA, Hagar J, Kashyap ML, Knopp RH, McKenney JM, Nash DT, Nash SD (2000) Extended-release niacin vs gemfibrozil for the treatment of low levels of high-density lipoprotein cholesterol. Arch Intern Med 160(8):1177–1184
Carlson LA (1963) Studies on the effect of nicotinic acid on catecholamine stimulated lipolysis in adipose tissue in vitro. Acta Med Scand 173:719–722
Lauring B, Taggart AKP, Tata JR, Dunbar R, Caro L, Cheng K, Chin J, Colletti SL, Cote J, Khalilieh S, Liu J, Luo W, MacLean AA, Peterson LB, Polis AB, Sirah W, Wu T, Liu X, Jin L, Wu K, Boatman PD, Semple G, Behan DP, Connolly DT, Lai E, Wagner JA, Wright SD, Cuffie C, Mitchel YB, Rader DJ, Paolini JF, Waters MG, Plump A (2012) Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med 4(148):115–124
Dobbins RL, Shearn SP, Byerly RL, Gao FF, Mahar KM, Napolitano A, Nachbaur GJ, Le Monnier de Gouville AC (2013) GSK256073, a selective agonist of G-protein coupled receptor 109A (GPR109A) reduces serum glucose in subjects with type 2 diabetes mellitus. Diabetes Obes Metab. doi:10.1111/dom.12132
Fulcher GR, Walker M, Catalano C, Agius L, Alberti KGMM (1992) Metabolic effects of suppression of nonesterified fatty acid levels with acipimox in obese NIDDM subjects. Diabetes 41(11):1400–1408
Kumar S, Durrington PN, Bhatnagar D, Laing I (1994) Suppression of non-esterified fatty acids to treat type A insulin resistance syndrome. Lancet 343(8905):1073–1074
Bajaj M, Suraamornkul S, Kashyap S, Cusi K, Mandarino L, DeFronzo RA (2004) Sustained reduction in plasma free fatty acid concentration improves insulin action without altering plasma adipocytokine levels in subjects with strong family history of type 2 diabetes. J Clin Endocrinol Metab 89(9):4649–4655
Worm D, Henriksen JE, Vaag A, Thye-Ronn P, Melander A, Beck-Nielsen H (1994) Pronounced blood glucose-lowering effect of the antilipolytic drug acipimox in noninsulin-dependent diabetes mellitus patients during a 3-day intensified treatment period. J Clin Endocrinol Metab 78(3):717–721
Metcalfe P, Johnston DG, Nosadini R (1981) Metabolic effects of acute and prolonged growth hormone excess in normal and insulin-deficient man. Diabetologia 20(2):123–128
Gill A, Johnston DG, Orskov H (1982) Metabolic interactions of glucagon and cortisol in man—studies with somatostatin. Metab Clin Exp 31(4):305–311
Mittelman SD, Bergman RN (2000) Inhibition of lipolysis causes suppression of endogenous glucose production independent of changes in insulin. Am J Physiol 279(3):630–637
Stralfors P, Bjorgell P, Belfrage P (1984) Hormonal regulation of hormone-sensitive lipase in intact adipocytes: identification of phosphorylated sites and effects on the phosphorylation by lipolytic hormones and insulin. Proc Natl Acad Sci USA 81(11):3317–3321
Frayn KN, Shadid S, Hamlani R, Humphreys SM, Clark ML, Fielding BA, Boland O, Coppack SW (1994) Regulation of fatty acid movement in human adipose tissue in the postabsorptive-to-postprandial transition. Am J Physiol Endocrinol Metab 266(3):308–317
Sadur CN, Eckel RH (1982) Insulin stimulation of adipose tissue lipoprotein lipase. Use of the euglycemic clamp technique. J Clin Invest 69(5):1119–1125
Ahlström C, Peletier LA, Jansson-Löfmark R, Gabrielsson J (2011) Feedback modeling of non-esterified fatty acids in rats after nicotinic acid infusions. J Pharmacokinet Pharmacodyn 38(1):1–24
Ahlström C, Peletier LA, Gabrielsson J (2011) Quantitative analysis of rate and extent of tolerance of biomarkers: application to nicotinic acid-induced changes in non-esterified fatty acids in rats. Eur J Pharm Sci 44(3):250–264
Ahlström C, Peletier LA, Gabrielsson J (2013) Challenge of a mechanistic feedback model describing nicotinic acid-induced changes in non-esterified fatty acids in rats. J Pharmacokinet Pharmacodyn 40(4):497–512
Levy G (1993) Integration of pharmacokinetics, pharmacodynamics and toxicokinetics in rational drug development, the case for preclinical pharmacodynamics. Plenum, New York
Zucker L, Zucker T (1961) Fatty, a new mutation in the rat. J Hered 52:275–278
Shum L, Jusko WJ (1984) Theophylline disposition in obese rats. J Pharmacol Exp Ther 228(2):380–386
Jaber LA, Ducharme MP, Halapy H (1996) The effects of obesity on pharmacokinetics and pharmacodynamics of glipizide in patients with non-insulin-dependent diabetes mellitus. Ther Drug Monit 18(1):6–13
Cheymol G (2000) Effects of obesity on pharmacokinetics: implications for drug therapy. Clin Pharmacokinet 39(3):215–231
Cortinez LI, Anderson BJ, Penna A, Olivares L, Munoz HR, Holford NHG, Struys MMRF, Sepulveda P (2010) Influence of obesity on propofol pharmacokinetics: derivation of a pharmacokinetic model. Br J Anaesth 105(4):448–456
Hanley MJ, Abernethy DR, Greenblatt DJ (2010) Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet 49(2):71–87
Danhof M, Levy G (1984) Kinetics of drug action in disease states. I. Effect of infusion rate on phenobarbital concentrations in serum, brain and cerebrospinal fluid of normal rats at onset of loss of righting reflex. J Pharmacol Exp Ther 229(1):44–50
Rohner-Jeanrenaud F, Proietto J, Ionescu E, Jeanrenaud B (1986) Mechanism of abnormal oral glucose tolerance of genetically obese fa/fa rats. Diabetes 35(12):1350–1355
McCaleb ML, Sredy J (1992) Metabolic abnormalities of the hyperglycemic obese Zucker rat. Metab Clin Exp 41(5):522–525
Zucker LM, Antoniades HN (1972) Insulin and obesity in the Zucker genetically obese rat “fatty”. Endocrinology 90(5):1320–1330
Martin RJ, Wangsness PJ, Gahagan JH (1978) Diurnal changes in serum metabolites and hormones in lean and obese Zucker rats. Horm Metab Res 10(3):187–192
Tannenbaum GS, Lapointe M, Gurd W, Finkelstein JA (1990) Mechanisms of impaired growth hormone secretion in genetically obese Zucker rats: roles of growth hormone-releasing factor and somatostatin. Endocrinology 127(6):3087–3095
Terrettaz J, Assimacopoulos-Jeannet F, Jeanrenaud B (1986) Severe hepatic and peripheral insulin resistance as evidenced by euglycemic clamps in genetically obese fa/fa rats. Endocrinology 118(2):674–678
Eaton RP, Conway M, Schade DS (1976) Endogenous glucagon regulation in genetically hyperlipemic obese rats. Am J Physiol 230(5):1336–1341
Martin RJ, Gahagan JH (1977) The influence of age and fasting on serum hormones in the lean and obese-Zucker rat. Proc Soc Exp Biol Med 154(4):610–614
Popovic V, Popovic P (1960) Permanent cannulation of aorta and vena cava in rats and ground squirrels. J Appl Physiol 15(4):727–728
Isaksson C, Wallenius K, Peletier LA, Toresson H, Gabrielsson J (2009) Turnover modeling of non-esterified fatty acids in rats after multiple intravenous infusions of nicotinic acid. Dose-Response 7(3):247–269
van Schaick EA, Zuideveld KP, Tukker HE, Langemeijer MWE, Ijzerman AP, Danhof M (1998) Metabolic and cardiovascular effects of the adenosine A1 receptor agonist N6-(p-sulfophenyl)adenosine in diabetic Zucker rats: influence of the disease on the selectivity of action. J Pharmacol Exp Ther 287(1):21–30
Bray GA (1977) The Zucker-fatty rat: a review. Fed Proc 36(2):148–153
Schonfeld G, Pfleger B (1971) Overproduction of very low-density lipoproteins by livers of genetically obese rats. Am J Physiol 220(5):1178–1181
Schonfeld G, Felski C, Howald MA (1974) Characterization of the plasma lipoproteins of the genetically obese hyperlipoproteinemic Zucker fatty rat. J Lipid Res 15(5):457–464
Kato R (1977) Drug metabolism under pathological and abnormal physiological states in animals and man. Xenobiotica 7:25–92
Levy G (1977) Pharmacokinetics in renal disease. Am J Med 62(4):461–465
Breimer DD (1987) Pharmacokinetics in liver disease. Pharm Weekbl Sci Ed 9(2):79–84
Frayn KN, Williams CM, Arner P (1996) Are increased plasma non-esterified fatty acid concentrations a risk marker for coronary heart disease and other chronic diseases? Clin Sci 90(4):243–253
Rowland, Tozer (1995) Clinical pharmacokinetics, concepts and applications. Williams and Wilkins, Baltimore
Vaag AA, Beck-Nielsen H (1992) Effects of prolonged acipimox treatment on glucose and lipid metabolism and on in vivo insulin sensitivity in patients with non-insulin dependent diabetes mellitus. Acta Endocrinol (Copenh) 127(4):344–350
Garg A, Grundy SM (1990) Nicotinic acid as therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. J Am Med Assoc 264(6):723–726
Thadani U, Fung HL, Darke AC, Parker JO (1982) Oral isosorbide dinitrate in angina pectoris: comparison of duration of action and dose-response relation during acute and sustained therapy. Am J Cardiol 49(2):411–419
Jamrozik K, Fowler G, Vessey M, Wald N (1984) Placebo controlled trial of nicotine chewing gum in general practice. Br Med J 289(6448):794–797
Parker JO, Fung HL (1984) Transdermal nitroglycerin in angina pectoris. Am J Cardiol 54(6):471–476
Jordan RA, Seth L, Casebolt P (1986) Rapidly developing tolerance to transdermal nitroglycerin in congestive heart failure. Ann Intern Med 104(3):295–298
Elkayam U, Kulick D, McIntosh N (1987) Incidence of early tolerance to hemodynamic effects of continuous infusion of nitroglycerin in patients with coronary artery disease and heart failure. Circulation 76(3):577–584
Porchet HC, Benowitz NL, Sheiner LB (1988) Pharmacodynamic model of tolerance: application to nicotine. J Pharmacol Exp Ther 244(1):231–236
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Steady state response
The steady state values for the response R and the moderators M i (i = 1,…, N) of the feedback model, formulated in Eq. (3) and (4), are given by:
where R ss is the unique solution of the equation:
in which
and R 0 is the baseline response. There is no explicit solution to Eq. (11).
For the obese animals, k cap could not be estimated and was therefore fixed to zero throughout the analysis. Equation (11) can then be simplified and solved for R SS :
Response at baseline
At baseline, I(C p ) = 1, and Eq. (11) can be expressed as:
For the obese animals where k cap = 0, Eq. (14) can then be simplified and solved for R 0 :
For the normal rats k cap is assumed to be small compared to the other terms in Eq. (14), and the baseline can therefore be approximated according to Eq. (15).
Steady state response at IC 50
Assuming that I max = 1, the inhibitory drug mechanism function at IC 50 is equal to 0.5. For the obese rats, Eq. (13) can be expressed as:
If k cap for the normal animals is assumed to be smaller than the other terms in Eq. (11), the steady state response at IC 50 for the normal rats can be approximated by Eq. (16), although this will slightly underestimate the value of R SS .
Steady state response at high NiAc concentrations
As I max = 1, it follows from the definition of I(C p ) (Eq. 2) that \( I(C_{p} ) \to 0 \) as \( C_{p} \to \infty \). This implies that:
For the obese animals where k cap = 0 it follows that:
For the normal animals, the sigmoidicity factor γ is equal to 2.2. At NiAc concentrations higher than 0.5 μmol L−1, \( IC_{50}^{\gamma } \ll C_{p}^{\gamma } \) and I(C p ) ≈ 0. Equation (17) is therefore valid at NiAc concentrations of 0.5 μmol L−1 and higher.
For the obese animals, the sigmoidicity factor γ is equal to 0.35 and \( IC_{50}^{\gamma } \) will then extensively affect I(C p ) at NiAc concentrations far above 1,000 μmol L−1. The steady state response \( R_{SS,obese} \) will therefore not approach zero within a therapeutic concentration interval. The steady state response \( R_{SS,obese} \) at high NiAc concentrations is still described by Eq. (13).
Rights and permissions
About this article
Cite this article
Ahlström, C., Kroon, T., Peletier, L.A. et al. Feedback modeling of non-esterified fatty acids in obese Zucker rats after nicotinic acid infusions. J Pharmacokinet Pharmacodyn 40, 623–638 (2013). https://doi.org/10.1007/s10928-013-9335-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-013-9335-z