
Abstract
Enantioselective pharmacokinetics and absorption of eflornithine in the rat was investigated using population pharmacokinetic modeling and a modified deconvolution method. Bidirectional permeability of l- and d-eflornithine was investigated in Caco-2 cells. The rat was administered racemic eflornithine hydrochloride as a single oral dose [40–3,000 mg/kg bodyweight (BW)] or intravenously (IV) (100–2,700 mg/kg BW infused over 60–400 min). Serial arterial blood samples were collected and l- and d-eflornithine were quantitated with a previously published chiral bioanalysis method. The D:L concentration ratio was determined in rat faeces. Intravenous l-and d-eflornithine plasma concentration–time data was analyzed using population pharmacokinetic modeling and described with a 3-compartment pharmacokinetic model with saturable binding to one of the peripheral compartments. Oral plasma concentration–time data was analyzed using a modified deconvolution method accounting for nonlinearities in the eflornithine pharmacokinetics. Clearance was similar for both enantiomers (3.36 and 3.09 mL/min). Oral bioavailability was estimated by deconvolution at 30 and 59 % for l- and d-eflornithine. The D:L concentration ratio in feces was 0.49 and the Caco-2 cell permeability was similar for both enantiomers (6–10 × 10−8 cm/s) with no evident involvement of active transport or efflux. The results presented here suggest that the difference in the bioavailability between eflornithine enantiomers is caused by a stereoselective difference in extent rather than rate of absorption. The presented modified deconvolution method made it possible to account for the non-linear component in the suggested three-compartment pharmacokinetic model thus rapidly estimating eflornithine oral bioavailability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Eflornithine (DL-α-difluoromethylornithine, DFMO) is an irreversible inhibitor of ornithine decarboxylase (ODC), the enzyme responsible for polyamine biosynthesis [1, 2]. Polyamines are required for optimal cell growth both in bacteria and eukaryotic cells. Eflornithine was initially developed as a possible cancer treatment but never marketed for that indication. However, lately it has again been proposed for treatment of colon cancer [3–7].
During the early 1980’s eflornithine was found to be trypanostatic and it was suggested to be efficacious for treatment of Human African Trypanosomiasis (HAT), also known as African sleeping sickness [2]. HAT is a parasitic disease with a near 100 % mortality if left untreated [8]. It is estimated that 60 million people are at risk of being infected in sub-Saharan Africa, and the disease has been ranked as the world’s third most important parasitic disease when adjusted for life years lost [9]. HAT disease progression is divided into two stages; an early, haemolymphatic, stage and a late, encephalitic, stage. The early stage is characterized by diffuse influenza like symptoms and in the late encephalitic stage, once the parasite has entered into the central nervous system, an array of neurological symptoms occur that finally leads to death [9]. For the treatment of late-stage HAT, only a few drugs are available. The most commonly used treatments depend on either melarsoprol or eflornithine [10]. The arsenical drug melarsoprol is administered intravenously, known to cause severe side effects and recently parasite resistance has emerged [11]. Eflornithine-based treatment is recommended as first-line against late-stage HAT. Phase III clinical studies have shown that intravenous eflornithine combined with oral nifurtimox is a promising new treatment [12, 13]. This combination was recently adapted by the WHO [14]. The main problem with eflornithine treatment is the complicated mode of administration, consisting of intravenous infusions of 100–200 mg/kg BW, every 6th to 12th h for 7–14 days. Consequently, numerous patients are left untreated [12, 13, 15]. A simplified mode of administration, preferably oral, would enable more patients to gain access to treatment but attempts to develop an oral treatment has so far failed [15].
Eflornithine is administered as a racemic mixture and it has been shown that the enantiomers display different in vitro potency. The l-enantiomer has up to a 20-fold higher affinity for the target enzyme, ODC [1]. The l-enantiomer also appears to be more potent in cultured Trypanosoma brucei parasites (R. Brun, Swiss Tropical Institute, personal communication). The differing potency of the enantiomers suggests that it is primarily the l-form that elicits the trypanostatic effect. When considering alternative modes of administration it is therefore crucial to assess exposure of enantiomers separately.
The amino acid like drug eflornithine is a small (MW: 182.2 g/mol) hydrophilic compound (logP = −2.31) and does not bind significantly to plasma proteins [16]. Clinical non-stereoselective pharmacokinetics of eflornithine display a low volume of distribution of 0.2–0.8 L/kg BW, a low clearance of 1.2–2 mL/min/kg BW, of which 80 % is composed of renal clearance and a reported bioavailability of 50–100 % [16–18]. Eflornithine has no identified metabolites. There is previously only one peer-reviewed publication regarding the stereoselective pharmacokinetics of eflornithine where the pharmacokinetics after single oral and intravenous administration in rat was studied [19]. The study revealed that, after oral administration of a racemic mixture, the plasma concentrations of the l-enantiomer were approximately half compared to the d-enantiomer. In contrast, after intravenous administration enantiomer plasma concentrations were similar up to the last sampling point, 2 h after dose. It was also found that the absorption was non-linear at the studied doses. The difference in exposure after oral administration was suggested to be a consequence of stereoselective absorption over the gastrointestinal tract. However, due to the low sensitivity of the stereoselective bioanalytical method at hand, the dose levels used were high [20]. Racemic oral and intravenous doses of 750–3,000 and 375–1,000 mg/kg BW, was given. This compared to doses of 100–200 mg/kg BW two to four times daily given to patients. In addition, enantiomeric data was sparse preventing a detailed analysis of the stereoselective pharmacokinetics. A recently developed bioanalytical method enabled a more thorough investigation of the stereoselective pharmacokinetics of eflornithine at more clinically relevant doses [21].
The overall aim of the present study was to characterize the enantioselective population pharmacokinetics of eflornithine after intravenous infusion to the rat and to deconvolute rat oral plasma concentration–time data with a modified deconvolution method, thus providing an increased mechanistic understanding of the stereoselective eflornithine absorption. Eflornithine was studied after single oral administration and intravenous infusions to the rat covering a wide dose range (40–3,000 mg/kg BW) to detect potential non-linearities. Presystemic enantiomeric metabolism was investigated by quantitation of eflornithine enantiomers in feces and bidirectional permeability was investigated in Caco-2 cell monolayers both in presence and absence of a Pgp-inhibitor.
Materials and methods
Chemicals
Eflornithine hydrochloride monohydrate (MW 236.65 g/mol) was kindly donated by WHO/TDR (Geneva, Switzerland). Isoflurane (Forene; Abbot Scandinavia AB, Solna, Sweden) and heparin (Leo Pharma AB, Malmö, Sweden) were obtained from Apoteket AB (Sweden). All chemicals were of analytical grade, and all solvents were of high-performance liquid chromatography (HPLC) grade.
Animals
Male Sprague–Dawley rats (Charles River, Germany) weighing 300–365 g (average weight 338 g) were acclimatized for at least 5 days after arrival at a certified animal facility (Experimental Biomedicine at University of Gothenburg, Gothenburg, Sweden). The animals were housed under controlled environmental conditions (12-h light–dark cycle at 25–27 °C and 60–65 % humidity). Four rats were kept in each cage prior to surgery and thereafter were kept separately. Food (Harlan, USA, B&K Feeds Ponca City OK USA) and tap water were available ad libitum prior to and after surgery. All invasive experiments were performed during the light phase of the cycle. Blood samples were however collected during both cycles. The study was approved by the Ethics Committee for Animal Experiments, Göteborg, Sweden (255/2005).
Animal surgery
The animals were anesthetized by inhalation of isoflurane (2.9–3.7 % in air). Independent of route of administration, the left jugular vein was catheterized using MRE040 1.02-mm-outer-diameter (OD), 0.64-mm-inner-diameter (ID) tubing and the right carotid artery were catheterized using PE-50 0.96-mm OD, 0.58-mm-ID tubing (AgnThos, Lidingö, Sweden), prefilled with 100 IU/ml of heparin in saline solution. Both catheters were tunnelled subcutaneously to emerge at the back of the neck. Catheters were kept patent with heparinized saline solution (20 IU/ml) between sampling to prevent blood clotting. All animals were allowed to recover over night after surgery. Food was removed 6 h prior to dose administration and replaced 6 h after start of administration.
Experimental in vivo design
Eflornithine hydrochloride was administered either orally or as intravenous infusion. The rats were randomly divided into dose groups. Dose levels for oral administration ranged from 40 to 3,000 mg/kg BW of racemic eflornithine hydrochloride (Table 1). Solutions for oral administration were prepared by dissolving dry eflornithine hydrochloride powder (crystals) in saline solution. Intravenous infusions ranged from 100 to 2,700 mg/kg BW over 60–400 min. Infusions were carried out to resemble the oral plasma concentration–time profile (Table 1). Eflornithine solutions for intravenous administration were prepared likewise but pH was adjusted to 7.2 with sodium hydroxide solution.
From every fourth rat a plasma sample was taken prior to dose administration to ensure that endogenous compounds did not chromatographically interfere with eflornithine enantiomers. After drug administration, between 7 and 13 blood samples were collected for determination of eflornithine enantiomers in plasma.
Oral doses were administered by gavage at a volume of 10 mL/kg BW for each dose level (Table 1). Intravenous infusions were administered at a volume of 10 mL/kg BW, with durations varying between 60–400 min, and were designed to resemble the plasma concentration profile after oral administration (Table 1). During infusions, rats were connected to a balancing arm and swivel, ensuring they were freely moving. After the end of infusion, rats were disconnected from the balancing arm.
Feces fractions were collected from 11 (1–4 rats per dose level) rats receiving eflornithine orally for determination of the l- to d-eflornithine ratio.
Caco-2 cell studies
Bidirectional transport studies were performed on Caco-2 cell monolayers according to a previously published method [22]. Caco-2 cells (ATCC, passage 94-100) were seeded on cell culture inserts maintained at 10 % CO2 in an incubator with a humidified atmosphere at 37 °C. Cells were seeded on polycarbonate cell culture inserts (4.4 × 105 cells per cm2, Transwell system, diameter 12 mm, pore size 0.4 μm, Corning Costar, The Netherlands) and were allowed to differentiate for 24–25 days (1 days in DMEM supplemented with 10 % fetal calf serum, 1 % nonessential amino acids, and PEST). The mean transepithelial resistance values of the monolayers were 255 ± 16 (SD) Ω cm2 (n = 9 filters) before the experiment and decreased to 217 Ω cm2 after the transport studies. Experiments were performed in Hank’s balanced salt solution (HBSS) at a pH of 7.4 on both the apical and basolateral side. During transport experiments, samples were taken at regular time intervals (0, 40, 80, 120 min) and replaced by an equivalent volume of pre-tempered HBSS.
Racemic eflornithine was used in the experiments, corresponding to donor enantiomeric concentrations of 0.75 and 12.5 mM. Eflornithine was studied in both apical-to-basolateral and basolateral-to-apical direction. Pgp-inhibition studies in the presence of GF120918 (0.01 mM) was done at eflornithine enantiomer concentration of 0.75 mM. To ensure that polycarbonate cell culture insert were not a barrier for eflornithine, inserts in the absence of Caco-2 cells were incubated at a concentration of 12.5 mM of each enantiomer. The apparent permeability (Papp) were calculated according to Papp = dQ/dt × 1/(A × c 0), where dQ/dt is the steady-state flux (μmol/s), A is the surface area of the filter (cm2), and C 0 is the initial concentration in the donor chamber (μM).
l- and d-eflornithine determinations
l- and d-eflornithine were quantitated in plasma according to a previously described method based on precolumn derivatization and liquid chromatography with UV detection [21]. Prior to quantitation of enantiomers in the rat plasma samples it was ensured that human plasma could be used for calibrators and quality controls. Rat and human plasma were prepared at concentrations of 1.5, 3.75, 25, 125, 500 and 2,000 μM of each enantiomer and analyzed in triplicate at each level for both species. For the l-enantiomer, the mean (±standard error) slope and intercept were 1.028 ± 0.019 and 5.17 ± 81.7, respectively. For the d-enantiomer the mean slope and intercept were 1.030 ± 0.021 and 0.307 ± 94.5. The slope and intercepts did not deviate significantly from 1 and 0 (n = 18, t test; P > 0.95). The corresponding peak area for rat and human plasma were compared at each concentration level and enantiomer. The mean ratio between rat and human were 1.14 (n = 18, 95 % CI: 0.88–1.39) and 0.99 (n = 18, 95 % CI: 0.96–1.03), for l- and d-eflornithine. A calibration curve, prepared in human plasma, ranging from 1.5 to 1,250 μM of each enantiomer was used to determine eflornithine enantiomers. Quality control samples at three levels (3, 400, 1000 μM of each enantiomer) were analyzed in triplicates with each analytical assay. Accuracy and precision at each analytical assay were within 15 %.
Quantitation of eflornithine enantiomers in HBSS did not require sample preparation and was directly injected for analysis. Two calibration curves were prepared in HBSS: one ranging from 0.0127 to 1.03 μM (low calibration curve) and one range from 3.086 to 750 μM (high calibration curve) of each enantiomer. To increase sensitivity of the bioanalytical method in HBSS, detection of the low calibration curve was done with a Jasco 821-FP Intelligent Spectrofluorometer (Jasco, Tokyo, Japan) set at wavelength 340/440 nm (excitation/emission). The high calibration curve was detected with a Shimadzu SPD 10-A UV–Vis detector (Shimadzu Corp., Kyoto, Japan). The back calculated concentrations never deviating by more than 7 % from the nominal values. Caco-2 cell samples with a concentration higher than 750 μM were diluted in HBSS so they were within the range of the calibration curve.
The reference curve for feces was prepared by collecting feces from four rats not exposed to eflornithine. The feces was pooled and divided into six separate tubes and then diluted (4 droppings in 2 mL solution) with either blank deionized water, stock solution of 5, 100, 500, 1000, or 3000 μM of racemic eflornithine. These samples were used as a reference to ensure that the peak area ratio of l- and d-eflornithine was equivalent at the mentioned concentrations and were analyzed in the same analytical run as the rat samples.
The collected feces from rats administered eflornithine, was diluted with deionized water (4 droppings in 2 mL) and incubated at room temperature for 30 min under gentle agitation, simultaneously with the aforementioned references. Samples were then mixed with ice-cold methanol (2 mL) and stored at 8 °C for 30 min. Tubes were centrifuged (6 min, 1,770×g) and the supernatant was transferred to new tubes and centrifuged for an additional 10 min at 12,000×g. The supernatant was transferred to new tubes and evaporated to dryness at 50 °C, under a gentle stream of air. Dried samples were redissolved in 200 μL deionized water and 75 μL of the solution was transferred to injection vials and placed in the autoinjector for analysis. Rat feces samples with a higher peak area compared to the highest concentration reference were diluted (1:200 vol:vol) in deionized water and reanalyzed.
Pharmacokinetic analysis
The eflornithine plasma concentration–time data were analyzed in two steps. The intravenous data for l- and d-eflornithine was modeled with a population approach using nonlinear mixed effects modeling as implemented in NONMEM version VII (Icon Development Solutions, Ellicott City, Maryland, USA) [23]. The obtained population IV pharmacokinetic parameters from the modeling were in a second step used to deconvolute oral plasma concentration–time data to obtain bioavailability [24].
In the first step, several pharmacokinetic models were tested and model selection was based on mechanistic plausibility, parameter estimate precision, the objective function value (OFV) and for the suggested model (Fig. 1), a bootstrap (500 samples) was done to assess parameter precision, (%RSE, Table 2). Diagnostic plots including visual predictive check (VPC) (500 replicates) for the suggested model was also included in model discrimination. Additionally, discrimination between two nested models could be achieved by using the log-likelihood ratio since the difference in the OFV between the full and reduced models is approximately Chi square distributed. Decreases in the OFV for FOCE of 3.84 and 6.63 between two nested models (1 degree of freedom) implied the model with the smallest OFV being statistically superior (P < 0.05 and P < 0.01, respectively). In the parameter estimation, data were simultaneously fitted by the first-order conditional estimation (FOCE) method [23]. Individual parameter values were obtained as empirical Bayes estimates. A log-normal distribution was assumed for the inter-individual variability of the parameters (CL, V c , V t , Q), and a proportional residual error model was used. Model files and graphics were handled using the R based programs Xpose4 and PsN [25, 26].

Schematic illustration of the selected pharmacokinetic model. The model describes the l- and d-eflornithine disposition after intravenous infusion of racemic eflornithine. Pharmacokinetic parameters are defined in Table 2
The model that best described the intravenous plasma concentration–time data was a three-compartment pharmacokinetic model, where the binding into one of the peripheral compartments was saturable (Fig. 1) [27, 28].
The rate of change of concentration of drug in the central (Cp) and the first peripheral (Ct) compartments were defined as
and
where In(t) is the input of drug into the central compartment. The rate of change in concentration within the second peripheral compartment with saturable tissue binding was given as
where C b is the drug concentration in the second peripheral binding compartment.
Secondly, the in vivo stereoselective absorption of eflornithine was characterized. Plasma systemic input rate from gut for each experiment was deconvoluted from oral data, using population PK parameters obtained from the intravenous data. Since the system inferred from intravenous data was shown to contain a nonlinear component, ordinary deconvolution methodology could not be applied. Instead, a modification of the ordinary model described by Verotta [29] was done as described below. For rats receiving eflornithine orally, a smoothing spline interpolation (smoothing parameter = 0.1) was generated for each individual plasma concentration time-profile [30]. For each time series, the input rate [In in Eq. (1)] was incrementally identified by integrating the ordinary differential equations with small Euler steps (0.5 min) enforcing the concentration to match the corresponding smoothed concentration curve. For 14 of the 52 time-series in total (26 experiments, two enantiomers) the smoothing spline was occasionally negative in the concentration decline phase. To ensure positive values, the later parts of such time series were replaced by a smoothing spline interpolation derived on a logarithmic scale (followed by exponentiation). To investigate the robustness of the approach with respect to smoothing parameter, the analysis was repeated with smoothing parameters 0.01 and 1, reaching the same conclusions. To explore whether the Euler step was sufficiently small, the system was integrated with standard numerical methods (Stiff Matlab solver ode15 s; MathWorks, Inc) using interpolated values from the deconvoluted systemic input rate and confirming that the smoothed curve was retrieved.
Bioavailability was estimated by integrating the individual systemic input rate-time profiles up to the last observation. Maximum systemic input rate and time to reach maximum systemic input rate were obtained from the individual deconvoluted input rate-time profiles. The obtained parameters were summarized as mean values and 95 % confidence intervals (Table 3). A non-overlap of the 95 % confidence intervals for the parameters between different dose levels and enantiomers was interpreted as a significant difference.
Results
Plasma pharmacokinetics after intravenous infusions and oral administration
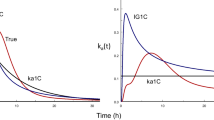
The lower limit of quantitation (LLOQ) for the bioanalysis method was 0.75 μmol/L for each enantiomer in plasma, 3.5 % of samples were below LLOQ and therefore omitted from the pharmacokinetic analysis. The three-compartment model with linear elimination and a non-linear binding component to one of the peripheral compartments was selected as the final model and could adequately describe the intravenous data (Figs. 1, 2, 3; Table 2). Separating parameters R max , k on and k off between the enantiomers did not significantly improve the model fit and were therefore estimated as identical for both l- and d-eflornithine. Interindividual variability (IIV) was estimated for clearance, central and peripheral volume of distribution as well as for intercompartmental clearance.

Goodness-of-fit plots for the final model after intravenous infusion of racemic eflornithine. The measured l-eflornithine and d-eflornithine concentrations were plotted versus the population predicted (a) and individual predictions (b) and conditional weighted residuals (CWRES) versus time after dose (c) and CWRES versus population predicted concentration (d)

Visual predictive check (VPC) with 95 % prediction interval of the model for l-and d-eflornithine. Observed data is depicted as circles. The solid black line is the model predicted median concentration with the corresponding 95 % confidence interval (CI) for the prediction in shaded grey. The dotted black line corresponds to the median of the observations. Doses levels correspond to racemic eflornithine doses administered to the rat
After oral administration of racemic eflornithine, time to reach maximum plasma concentration (Tmax) and time to reach maximum systemic input rate were similar for both enantiomers (Fig. 4; Table 3). The mean oral bioavailability of l-eflornithine (30 %) was approximately half compared to that for d-eflornithine (59 %) (Table 3). The maximum systemic input rate and time to reach maximum systemic input rate for eflornithine enantiomers were constant up to a racemic dose level of 400 mg/kg BW, above which systemic input was delayed (Maximum systemic input rate/dose and Time to reach maximum systemic input rate, Table 3). A slight increase in bioavailability was observed at the two highest dose levels (1,200 and 3,000 mg/kg BW), although not significantly different (95 % confidence intervals overlapped). The systemic input rate-time profiles were similar for both l- and d-eflornithine, however differing in magnitude (Fig. 4).

Plasma systemic input rate-time profiles. Mean systemic input rate from gut versus time after oral administration of 40, 150, 400, 1200, and 3000 mg/kg BW of racemic eflornithine. Heavy dashed and solid lines represent the mean input rate-time profiles for l- and d-eflornithine, and the thin lines the corresponding 95 % confidence intervals. Mean systemic input rate-time profiles shown for up until last sampling time where n = 5 for all profiles
Feces d- to l-eflornithine ratios
Feces spiked with racemic eflornithine gave a reference median D:L peak area ratio of 1.03 (range: 0.98–1.15, n = 6). For the rats being administered eflornithine orally the mean (±standard error) D:L peak area ratio was 0.49 ± 0.03 (n = 11) and range of 0.3–0.6; thus the inverse ratio to that observed in plasma and for the bioavailability of each enantiomer (Table 3).
Bidirectional transport in Caco-2 cells
Eflornithine displayed a low permeability in Caco-2 cells at donor enantiomeric concentrations of 0.75 and 12.5 mM. The permeability rates for the enantiomers were comparable. An indication of efflux was observed but considering the overall low permeability and in relation to in vivo data this efflux rate was considered negligible. Inhibition of P-glycoprotein did not affect the permeability of eflornithine enantiomers at these concentrations (Table 4).
Discussion
Eflornithine elicit its trypanostatic effect on parasites lodged both in the haemolymphatic and in the central nervous system [31]. The enantiomers display differing in vitro potency towards the target enzyme ODC [1]. Achieving sufficient concentrations of the more potent l-eflornithine enantiomer, in plasma and the central nervous system is essential for an effective eradication of the parasite. An eflornithine CSF-concentration above 50 μmol/L is required for effective eradication of the parasite from the CNS [18]. Reaching sufficient in vivo eflornithine exposure depends on a complicated intravenous mode of administration (100–200 mg/kg BW two to four times daily for up to 14 days), corresponding to a steady state plasma concentration of approximately 600 μmol/L. If this mode of administration could be simplified, preferably by an oral dosage regimen, more patients would gain access to treatment. However, the limiting factor to developing an oral eflornithine treatment is likely gastrointestinal side effects and/or inadequate bioavailability at the presently administered doses [15]. Whether an oral dosage regimen could result in an increased systemic exposure depends highly on the mechanism that limits the gastrointestinal absorption of l-eflornithine. Possibly, if the stereoselective absorption is driven by an efflux mechanism, not present in Caco-2 cells, the transporter may be inhibited by a selective inhibitor rendering higher systemic exposure of eflornithine. Should absorption be primarily driven by an active transporter, lower doses (or l-eflornithine alone) given more frequently might increase the bioavailability hasty. Considering this, there was great relevance to further investigating the enantioselective and nonlinear absorption of eflornithine enantiomers. In agreement with a previous study, absorption of the enantiomers was stereoselective with the more potent enantiomer less well absorbed [19]. In contrast to the previous study, a wider dose range was used here, better reflecting clinically relevant doses [15, 19]. An oral dose given to the rat of 100 mg/kg BW, with regards to local expected drug concentration in the gut, would approximately correspond to the same dose in humans. In terms of systemic exposure, a racemic oral dose of 700 mg/kg BW to the rat resulted in a similar AUC to patients at steady state treated with 100 mg/kg BW orally [15]. This observed difference in dose level is caused by AUC being dependent on oral clearance, and the local gut concentration dependent on the gut volume.
The sensitive bioanalysis method used here, enabled richer data to be collected from each individual animal. Thus, for both the intravenous and oral data, a wide dose range was studied. Intravenous doses were administered as constant infusions at varying durations to mimic the oral absorption profile and to evaluate possibilities of nonlinear disposition pharmacokinetics. The intravenous data was adequately described with a three-compartment pharmacokinetic model with linear systemic elimination and with a saturable binding to one of the peripheral compartments (Fig. 1) [27, 28]. Enantioselective k on and k off was investigated, however the model fit was not significantly improved (no drop in OFV) when incorporating these two parameters. Therefore separate k on and k off were omitted and estimated commonly for l- and d-eflornithine. In line with previous observations, separating the k on and k off gave an indication of difference in target affinity favoring the l-enantiomer [1]. Predicted total amount of ODC in the rat from measured amount in rat liver were about one tenth of the R max estimated by modeling (Table 2). Therefore, it might not only be the binding to ODC that effects the distribution of eflornithine enantiomers [32].
Eflornithine binding to ODC has previously been investigated in vitro using recombinant human ODC, with a reported KD-value of 2.2 ± 0.4 μmol/L [1]. This in vitro KD-value was smaller than the KD-value derived from the estimated k on and k off presented here of 170 μmol/L. The large difference observed in target affinity could further be explained by the physiological factors not present in the in vitro system.
Ornithine decarboxylase is an enzyme crucial for polyamine biosynthesis in eukaryote cells, and hence cell proliferation. The presented 3-compartment model suggests distribution to a saturable binding compartment from the central compartment (Fig. 1). Previously, distribution to the saturable binding compartment was investigated from the peripheral compartment, however based on the model discrimination criteria presented here this did not render a superior model fit to the presented model and was omitted.
Clearance for l- and d-eflornithine (3.36 and 3.09 ml/min) was similar to the glomerular filtration rate in the rat (8.7 mL/min/kg BW) and the central volume of distribution (75 and 72 mL) was similar to the rat extracellular fluid volume (74 mL for a 250 g rat) [33, 34]. The decrease in CL and V with increasing doses as observed, but not shown, with noncompartmental analysis (NCA) may indicate target mediated drug disposition [28]. However the study design at hand did not allow for this to be confirmed. The visual predictive check (Fig. 3) show an underprediction by the model at low doses (40 mg/kg BW), though based on only three individuals. This underprediction might have been an artifact of the study design since sampling for the lower doses was taken during a shorter period of time.
To characterize the in vivo stereoselective absorption of eflornithine, plasma systemic input rate from gut was deconvoluted from oral plasma concentration–time data with the suggested pharmacokinetic model and corresponding IV population parameters. Ordinary deconvolution methods could not be applied since this method is based on linear pharmacokinetics [27]. Therefore a modification of the ordinary deconvolution method was done which allowed for deconvolution of oral non-linear pharmacokinetic data.
The deconvolution method offers a rapid way of accessing the absorption temporal profile, and estimate bioavailability. Particularly useful when absorption is non-linear, multiphasic and/or when prior information is not rich enough for mechanistic modeling. The computational time of the method is mainly determined by the required Euler step size. In general, highly nonlinear systems can be expected to require small step sizes corresponding to long execution times. In our case, the required step size (0.5 min; moderately nonlinear kinetics) resulted in a computational time in the order of seconds on a standard desktop computer.
In feces collected after oral administration, concentrations of the more potent l-enantiomer was on average two-fold higher compared to the d-enantiomer. This was the inverse concentration ratio to that observed in plasma and to the estimated different bioavailabilies presented here (Table 3). This strongly suggests that the enantioselective exposure after oral administration of racemic mixture is not a consequence of presystemic stereoselective metabolism.
The time to reach maximum systemic input rate (Table 3) and time to reach maximum plasma concentration (Tmax) in the plasma concentration–time profile (not shown but estimated by NCA) give similar values and was not significantly different between the enantiomers. Therefore, the enantiomers are expected to have similar absorption rates. The observed difference in bioavailability of 30 and 59 % for l- and d-eflornithine, would then be explained by a stereoselectivity in the extent of absorption, which is observed when integrating the systemic plasma input rate-time profile (Fig. 4). However, with increasing oral doses there was a significant delay in Tmax and a delay in time to reach the maximum systemic input rate (Fig. 4; Table 3). This peak shift combined with the systemic input rate not reaching a plateau (Fig. 4), suggests that the uptake over the gastrointestinal tract was not driven by a saturated active uptake or that the active uptake was masked by other physiological processes. In addition, if the saturable systemic input rate would be driven by an active transporter it would be expected that the bioavailability decrease with an increase in dose, which was not observed here.
The eflornithine enantiomers display similar permeability over the Caco-2 cell monolayer (Table 4). The relatively high donor concentrations might impact study of active transport in the apical-to-basolateral direction due to saturation of active transporters. There are also some Caco-2 cell transporters (such as SLC15A1 and SLCO2B1) that require a proton gradient (i.e. pH 6.5 on the apical and 7.4 on the basolateral side) to be active. As such the present Caco-2 cell study could not exclude the possibility of an active uptake over the gastrointestinal tract.
Considering structural similarity between eflornithine and ornithine, the absorption mechanism of eflornithine may resemble that of ornithine. Ornithine, like cysteine and other dibasic amino acids are absorbed via the heteromeric intestinal amino acid transporter b°,+ (SLC7A9) [35]. Absorption of ornithine has been studied in humans and it has been suggested that ornithine displays both a saturable and a passive non saturable absorption [36]. Contradicting eflornithine enantiomers being substrates for an amino acid transporter is that absorption of the d-enantiomer is favored over the l-enantiomer. Eflornithine has also structural similarities to the cationic amino acids arginine and lysine. Both amino acids show nonlinear absorption. In animal studies, arginine was predominantly transported over the intestinal membrane via a Na+-independent System y+ (SLC7A1) [37]. During human intestinal perfusion studies of arginine and lysine, at concentrations above 100 mM, absorption ceased and excessive secretion of water and electrolytes into the lumen occurred. This was suggested to be a nonspecific toxic reaction of arginine in the mucosa [35]. In the present study concentrations of eflornithine solution given orally to the rat ranged between 10 and 730 mM, which may have caused a similar toxic reaction to those of arginine and lysine at the two highest dose levels, thus leading to a delay in time to reach maximum systemic input rate (Table 3). In humans, gastrointestinal side effects has been reported in 10–39 % of eflornithine patients, with diarrhea being more common after oral administration [18]. The gastrointestinal side effects may be more related to the l-enantiomer as the relative amount of this enantiomer in feces after oral administration to the rat, was found be higher compared to the d-enantiomer.
Based on the present Caco-2 permeability data, the predicted fraction absorbed in man using SimCYP™ V. 8.00 (Simcyp Limited©, Sheffield, UK) was approximately 4 %, which is about tenfold lower to that observed in man. This underprediction suggests that the passive transcellular route of absorption is a relatively minor route of absorption for eflornithine and that there are additional mechanisms present. Previous reports have suggested that the paracellular route has a significant role in accurate prediction of absorption using in vitro screening techniques [38]. For Caco-2 cells, the paracellular pore radius is smaller than that observed in the small intestine in vivo and it has been suggested that this is one reason why Caco-2 cells underpredict fraction absorbed for compounds primarily absorbed through the paracellular pathway. The paracellular route of absorption has also been suggested to be limited to small hydrophilic molecules (MW < 250 g/mol), like eflornithine [39, 40].
Contradicting an eflornithine paracellular route of absorption is also the stereoselective process observed here. Though, the paracellular network is an intricate system consisting of numerous proteins forming the junctional complex [41]. Considering that eflornithine enantiomers have different affinity towards the target enzyme, ODC, it may be hypothesized that the l-form in general also have a higher affinity towards structures forming the tight junctions. It has previously been reported that eflornithine decreases mucosal ODC activity and polyamine expression in the junctional complex [42]. Should the l-enantiomer have a higher affinity towards structures in the paracellular network, compared to the d-enantiomer, this would result in lower paracellular permeability and subsequently a lower fraction absorbed of the l-enantiomer compared to the d-enantiomer.
The bioavailability for l- and d-eflornithine was estimated at 30 and 59 %. Thus, the L:D bioavailability ratio was 1:2, the inverse ratio to that observed in feces (2:1) and supporting an enantioselective extent of absorption between the enantiomers. It may then be hypothesized that the l-enantiomer could be prone to forming a chemical complex in the gut prior to absorption rendering larger, bulky, molecules that would be less well absorbed. However, the plausibility of this explanation renders further investigations.
In conclusion, the intravenous pharmacokinetics of l- and d-eflornithine was described with a 3-compartment pharmacokinetic model with linear elimination and saturable binding to one of the peripheral compartments. In order to characterize the in vivo stereoselective absorption of eflornithine, ordinary deconvolution methods could not be applied and was therefore modified to describe a system containing a non-linear component. Using this method it was concluded that the bioavailability of eflornithine was independent of dose. The bioavailability was estimated at 30 and 59 % for l- and d-eflornithine after oral racemic eflornithine doses (40–3,000 mg/kg BW) administered to the rat.
At doses above 400 mg/kg BW, both time to reach maximum systemic input rate and maximum systemic input rate normalized to dose were dose-dependent and a delay with increasing dose was observed, but with no significant difference between the enantiomers. A conclusive mechanism causing the observed enantioselectivity in bioavailability was not identified. However, a number of mechanisms could be excluded. Based on the present results, the stereoselective absorption is either driven by a stereoselective paracellular absorption mechanism, chiral chemical complex formation or by an active uptake in the gastrointestinal tract. To further mechanistically elucidate the stereoselective absorption, simultaneous modeling of the oral plasma concentration–time data along additional in vitro absorption studies could provide further information. In particular, stereoselective and dose dependent aspects of the delay in time to reach maximum systemic input rate requires further investigation.
References
Qu N, Ignatenko NA, Yamauchi P, Stringer DE, Levenson C, Shannon P, Perrin S, Gerner EW (2003) Inhibition of human ornithine decarboxylase activity by enantiomers of difluoromethylornithine. Biochem J 375(Pt 2):465–470
Bacchi CJ, Nathan HC, Hutner SH, McCann PP, Sjoerdsma A (1980) Polyamine metabolism: a potential therapeutic target in trypanosomes. Science 210(4467):332–334
Raul F (2007) Revival of 2-(difluoromethyl)ornithine (DFMO), an inhibitor of polyamine biosynthesis, as a cancer chemopreventive agent. Biochem Soc Trans 35(Pt 2):353–355
Ignatenko NA, Besselsen DG, Stringer DE, Blohm-Mangone KA, Cui H, Gerner EW (2008) Combination chemoprevention of intestinal carcinogenesis in a murine model of familial adenomatous polyposis. Nutr Cancer 60(Suppl 1):30–35
Gerner EW, Meyskens FL Jr, Goldschmid S, Lance P, Pelot D (2007) Rationale for, and design of, a clinical trial targeting polyamine metabolism for colon cancer chemoprevention. Amino Acids 33(2):189–195
McLaren CE, Fujikawa-Brooks S, Chen WP, Gillen DL, Pelot D, Gerner EW, Meyskens FL Jr (2008) Longitudinal assessment of air conduction audiograms in a phase III clinical trial of difluoromethylornithine and sulindac for prevention of sporadic colorectal adenomas. Cancer Prev Res (Phila Pa) 1(7):514–521
Mackenzie GG, Ouyang N, Xie G, Vrankova K, Huang L, Sun Y, Komninou D, Kopelovich L, Rigas B (2011) Phospho-sulindac (OXT-328) combined with difluoromethylornithine prevents colon cancer in mice. Cancer Prev Res (Phila) 4(7):1052–1060
Kennedy PG (2004) Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest 113(4):496–504
Kennedy PG (2008) The continuing problem of human African trypanosomiasis (sleeping sickness). Ann Neurol 64(2):116–126
Jacobs RT, Nare B, Phillips MA (2011) State of the art in African trypanosome drug discovery. Curr Top Med Chem 11(10):1255–1274
Docampo R, Moreno SN (2003) Current chemotherapy of human African trypanosomiasis. Parasitol Res 90(Supp 1):S10–S13
Balasegaram M, Young H, Chappuis F, Priotto G, Raguenaud ME, Checchi F (2008) Effectiveness of melarsoprol and eflornithine as first-line regimens for gambiense sleeping sickness in nine Medecins Sans Frontieres programmes. Trans R Soc Trop Med Hyg 103(3):280–290
Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, Ghabri S, Baudin E, Buard V, Kazadi-Kyanza S, Ilunga M, Mutangala W, Pohlig G, Schmid C, Karunakara U, Torreele E, Kande V (2009) Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Lancet 374(9683):56–64
Burri C (2010) Chemotherapy against human African trypanosomiasis: is there a road to success? Parasitology 137(14):1987–1994
Na-Bangchang K, Doua F, Konsil J, Hanpitakpong W, Kamanikom B, Kuzoe F (2004) The pharmacokinetics of eflornithine (alpha-difluoromethylornithine) in patients with late-stage T.b. gambiense sleeping sickness. Eur J Clin Pharmacol 60(4):269–278
Sanderson L, Dogruel M, Rodgers J, Bradley B, Thomas SA (2008) The blood–brain barrier significantly limits eflornithine entry into Trypanosoma brucei brucei infected mouse brain. J Neurochem 107(4):1136–1146
Haegele KD, Alken RG, Grove J, Schechter PJ, Koch-Weser J (1981) Kinetics of alpha-difluoromethylornithine: an irreversible inhibitor of ornithine decarboxylase. Clin Pharmacol Ther 30(2):210–217
Burri C, Brun R (2003) Eflornithine for the treatment of human African trypanosomiasis. Parasitol Res 90(Supp 1):S49–S52
Jansson R, Malm M, Roth C, Ashton M (2008) Enantioselective and nonlinear intestinal absorption of eflornithine in the rat. Antimicrob Agents Chemother 52(8):2842–2848
Malm M, Bergqvist Y (2007) Determination of eflornithine enantiomers in plasma, by solid-phase extraction and liquid chromatography with evaporative light-scattering detection. J Chromatogr B Analyt Technol Biomed Life Sci 846(1–2):98–104
Jansson-Lofmark R, Romsing S, Albers E, Ashton M (2010) Determination of eflornithine enantiomers in plasma by precolumn derivatization with o-phthalaldehyde-N-acetyl-l-cysteine and liquid chromatography with UV detection. Biomed Chromatogr 24(7):768–773
Hubatsch I, Ragnarsson EG, Artursson P (2007) Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc 2(9):2111–2119
Beal S (1986–2006) NONMEM users guide. In: Boeckmann A ICON Development Solutions, Ellicot City
Madden FN, Godfrey KR, Chappell MJ, Hovorka R, Bates RA (1996) A comparison of six deconvolution techniques. J Pharmacokinet Biopharm 24(3):283–299
Jonsson EN, Karlsson MO (1999) Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58(1):51–64
Lindbom L, Pihlgren P, Jonsson EN (2005) PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 79(3):241–257
Karlsson MO, Molnar V, Freijs A, Nygren P, Bergh J, Larsson R (1999) Pharmacokinetic models for the saturable distribution of paclitaxel. Drug Metab Dispos 27(10):1220–1223
Mager DE, Jusko WJ (2001) General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn 28(6):507–532
Verotta D (1996) Concepts, properties, and applications of linear systems to describe distribution, identify input, and control endogenous substances and drugs in biological systems. Crit Rev Biomed Eng 24(2–3):73–139
de Boor C (2001) Smoothing and least-squares approximation. In: Applied mathematical sciences 27—a practical guide to splines. Springer, New York, pp 211–214
Barrett MP, Boykin DW, Brun R, Tidwell RR (2007) Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. Br J Pharmacol 152(8):1155–1171
Seely JE, Poso H, Pegg AE (1982) Measurement of the number of ornithine decarboxylase molecules in rat and mouse tissues under various physiological conditions by binding of radiolabelled alpha-difluoromethylornithine. Biochem J 206(2):311–318
Lin JH (1995) Species similarities and differences in pharmacokinetics. Drug Metab Dispos 23(10):1008–1021
Davies B, Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10(7):1093–1095
Grimble GK (2007) Adverse gastrointestinal effects of arginine and related amino acids. J Nutr 137(6 Suppl 2):1693S–1701S
Cynober L (2007) Pharmacokinetics of arginine and related amino acids. J Nutr 137(6 Suppl 2):1646S–1649S
Pan M, Choudry HA, Epler MJ, Meng Q, Karinch A, Lin C, Souba W (2004) Arginine transport in catabolic disease states. J Nutr 134(10 Suppl):2826S–2829S discussion 2853S
Matsson P, Bergstrom CA, Nagahara N, Tavelin S, Norinder U, Artursson P (2005) Exploring the role of different drug transport routes in permeability screening. J Med Chem 48(2):604–613
Lennernas H (2007) Intestinal permeability and its relevance for absorption and elimination. Xenobiotica 37(10–11):1015–1051
Linnankoski J, Makela J, Palmgren J, Mauriala T, Vedin C, Ungell AL, Lazorova L, Artursson P, Urtti A, Yliperttula M (2010) Paracellular porosity and pore size of the human intestinal epithelium in tissue and cell culture models. J Pharm Sci 99(4):2166–2175
Chiba H, Osanai M, Murata M, Kojima T, Sawada N (2008) Transmembrane proteins of tight junctions. Biochim Biophys Acta 1778(3):588–600
Liu L, Guo X, Rao JN, Zou T, Xiao L, Yu T, Timmons JA, Turner DJ, Wang JY (2009) Polyamines regulate E-cadherin transcription through c-Myc modulating intestinal epithelial barrier function. Am J Physiol Cell Physiol 296(4):C801–C810
Acknowledgments
The provision of eflornithine by WHO/TDR is gratefully acknowledged. Dr Ina Hubatsch, Uppsala University, Uppsala Sweden, is acknowledged for her contribution to the Caco-2 cell assay.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johansson, CC., Gennemark, P., Artursson, P. et al. Population pharmacokinetic modeling and deconvolution of enantioselective absorption of eflornithine in the rat. J Pharmacokinet Pharmacodyn 40, 117–128 (2013). https://doi.org/10.1007/s10928-012-9293-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-012-9293-x