
Abstract
Green manufacturing and reducing our cultural dependency on petrochemicals have been the global interest currently, especially in the polyurethane industry segments. We report the fabrication of rigid polyurethane foams (RPUFs) and their flame-retardant property from hemp seed oil as an alternative to petrochemical-based polyols. The cold-pressed hemp-seed oil (HSO) was first oxidized to epoxidized triglyceride oils with acetic acid and hydrogen peroxide, followed by a ring-opening reaction with methanol to fabricate hemp bio-polyols. The formation of polyols was characterized using FT-IR, hydroxyl, and acid values. The bio-polyol was used in different proportions with commercial polyols and other foaming ingredients to produce rigid polyurethane foams via a one-step process. Dimethyl methylphosphonate (DMMP), triethyl phosphate (TEP), and expandable graphite (EG) were added during the foam preparation to improve flame retardancy. The produced foams were analyzed for their apparent density, mechanical properties, thermal degradation behavior, closed cell content, flammability, and cellular morphology. The effect of different flame retardants had a significant influence on the cellular structures, closed-cell content, density, and compressive strength of the polyurethane. A significant improvement in anti-flaming properties was observed as the neat HSO-based foam showed a burning time of 110 s and a weight loss of 82%, whereas 10 wt% of TEP displayed a reduced burning time and weight loss of 19 s and 5%, respectively. DMMP and EG-based RPUFs exhibited similar flame retardancy and mechanical properties relative to neat HSO-based foam. The results demonstrated in this work proposed a potential combination of bio-polyols and commercial polyols as a strategy to fabricate flame-retardant polyurethane foam for high-performance applications.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyurethane (PU) is used in many aspects of our daily lives due to its versatility. PU foam (rigid, semi-rigid, and flexible) production continues to increase globally due to its applications throughout different sectors including construction, furniture, packaging, electronics, coatings, footwear, adhesives, insulation foam, and aerospace [1, 2]. The polyurethane industry is often divided into foams (65%, including 40% rigid foams and 60% flexible foams), elastomers (12%), coatings (13%), adhesives (7%), and miscellaneous (3%) [3, 4]. Most of the polyurethane foams supplied in the market are made from petroleum byproducts i.e., non-renewable resources. Typically, polyols and diisocyanates derived petrochemically are the main constituents for the commercial production of PU products [3]. To reduce petroleum consumption, scientists and chemists are shifting their focus to utilizing safer and green alternative raw materials [5, 6]. Preparing polymers using renewable resources is crucial for both science and the economy [7, 8]. Petrochemical polyols can be effectively replaced by bio-polyols made from waste resources or plant oils for making PU foams [9,10,11,12]. Biobased polyol is obtained from the agriculture crop protein, wood, and vegetable oils such as canola, sunflower, hemp seed, and soybean [8, 9].
One of the suitable approaches is to use vegetable oil as a substitute for petroleum oil in the PU production process [9,10,11,12,13]. Vegetable oils have been a tempting replacement because of unsaturated double bonds and hydroxyl groups, which can be functionalized or directly utilized to prepare PU foams [8, 13, 14]. Also, they are cheap, widely available, and sustainable. Many vegetable oils, including soybean oil [15,16,17,18], linseed oil [17, 19], palm oil [20], castor oil [21, 22], and rapeseed oil [23], are well-known and are utilized to prepare bio-based polyols that may replace their petrochemical equivalents. Industrial hemp (Cannabis sativa L.), another oilseed crop, has significant promise in the chemical industry. Hemp is a remarkable crop with immense social and economic significance, given that it can be used to make food, textiles, plastics, paints, lighting oil, and cosmetic products [24, 25]. The hemp plant’s various sections are a good source of food and nutritional supplement elements. Hempseed is a great source of fiber, vitamins, minerals, and vital amino acids. Hemp sprouts are high in antioxidants, and hempseed oil is a good source of polyunsaturated fatty acids [24,25,26]. Although hemp seed oil is classified as edible oil, however, due to its low smoke point, it is not preferred for cooking. Hempseed oil (HSO) is obtained by cold-pressing the hemp seed [26]. HSO is distinguished by a very high content of linoleic acid (55–60%), and linolenic acid (17–35%). In comparison to sunflower oil (110–143 g I2/100 g), soybean oil (128–143 g I2/100 g), palm oil (44–58 g I2/100 g), and rapeseed oil (110–126 g I2/100 g), HSO has a higher iodine value ranging from 140 to 175 g I2/100 g [25, 26]. Epoxidized HSOs are being researched as starting materials for the synthesis of bio-based epoxy resins [27–29] and acrylate biopolymers, which are mostly used as coating materials for plastic, paper, and wood [30]. Surender et al. [31] epoxidized the oil and then produced HSO polyols by ring-opening reactions with butanol, ethanol, and water for synthesizing polyurethane elastomers. Further, there are no such reports on the synthesis of rigid polyurethane foams (RPUFs) containing HSO-based bio-polyols and their flame-retardant properties. RPUFs are highly combustible despite their versatility, therefore enhancing the flame resistance is essential to prevent dangerous fire situations that endanger people’s lives.
Herein, our current research investigates the development of rigid polyurethane foams from biobased hemp-seed polyol entirely synthesized from HSO. The transformation of HSO into an HSO-polyol involves an epoxidation reaction followed by oxirane ring-opening using methanol and tetrafluoroboric acid as catalysts. The use of additives (catalysts, surfactant) and the effect of different flame retardants on the final characteristics, and mechanical and functional properties of the foams are investigated. The final foams were subjected to scanning electron microscopy to assess their morphology and evaluated for apparent density, mechanical strength, closed cell content, and thermal stability. The focus of this work is to summarize the use of various flame retardants (FRs) on the in-situ preparation and flame retardancy properties of the RPUFs. Keeping environmental and safety hazards in principle, non-halogenated FRs like dimethyl methylphosphate (DMMP), triethyl phosphate (TEP), and expandable graphite (EG) were investigated as additives for flammability, mechanical strength, and morphology of the fabricated RPUFs. This feasibility research could serve as a foundation for future evidence-based product development, as it focuses on multi-component flame retardant systems that are almost ready for use.
Materials and Methods
Materials
Cold-pressed raw hemp seed oil (HSO) was happily provided by Midwest Hemp Technology from Kansas-grown hemp grains, extracted through CO2 processing. For the synthesis of the HSO-polyol, tetrafluoroboric acid (HBF4), Lewitt MP64, sodium sulfate, Amberlite IR 120 H, sodium chloride, methanol, hydrogen peroxide, acetic acid, and toluene were bought from Fisher Scientific, USA. Dibutyltin dilaurate (DBTDL) and 1,4-diazabicyclo [2.2.2] octane (DABCO) catalysts were acquired from Air Products (Allentown, PA, USA) for rigid foam preparation. Tegostab B-8404 (silicon surfactant) was obtained from Evonik (Parsippany, NJ, USA). Jeffol SG-522 and Rubinate M isocyanate were procured from Huntsman, Woodlands, USA. Dimethyl methyl phosphate (DMMP), triethyl phosphate (TEP), and expandable graphite (EG) as flame retardants were supplied by Sigma-Aldrich, USA. Deionized water used as blowing agents was obtained from Pittsburg Neighborhood Walmart.
Synthesis of Epoxidized Hempseed Oil (EHSO)
In this reaction, cold-pressed filtered HSO, toluene, and Amberlite resin were mechanically stirred in a 1000 ml three-necked round-bottom flask attached with a thermometer and a reflux condenser placed in a water bath following previously adopted routes [32]. The flasks were filled with a stoichiometric amount of HSO oil and Amberlite IR 120 H catalyst (15% by weight of oil), and the bath temperature was then adjusted to 65 °C. Then, acetic acid and hydrogen peroxide (0.5:1.5 molar ratio to double bond) were added. After continuously stirring for 30 min, the temperature increased to 70 °C and stirred again for 7 h. Following a room-temperature cooling of the mixture, the resin was filtered. After that, the oil was washed with 10% brine solution and distilled under a reduced pressure rotary to remove excess toluene to get the epoxidized hemp seed oil (EHSO).
Synthesis of Hempseed Polyols (HSPO)
The syntheses of bio-polyol were carried out in a 1000 ml reactor equipped with an oil bath, a mechanical stirrer, a dropping funnel, and a thermometer. Methanol and epoxidized HSO were mixed in a mole ratio of 7:1. Tetrafluoroboric acid, HBF4 (concentration of 50% water by weight) was added and the temperature was set at 70 °C. Then, the synthesized EHSO was added dropwise and the reaction was refluxed for an hour. To stop the hydrolysis, Lewatit MP 64 ion exchange resin was added after the liquid mixture had cooled. Following that, the resin was filtered and the excess solvents were removed by rotary evaporation under reduced pressure. The characterization of polyol formation was done through different confirmatory tests such as hydroxyl value, molecular weight via gel permeation chromatography (GPC), and molecular structure by Fourier-transform infrared spectroscopy (FT-IR). The synthetic route is illustrated in Fig. 1.

Schematic diagram for the synthesis and preparation of HSPO and HSO-based RPUFs
Preparation of HSO-Based Rigid Polyurethane Foams
The RPUFs were prepared using a one-pot method and formed inside a disposable plastic cup mold, as displayed in Fig. 1. The compositions of hempseed-oil-based rigid polyurethane foams are tabulated in Table 1. Three sets of foams with variable DMMP, TEP, and EG contents were prepared. F-1 to F-6 sample stands for foam formulation representative of the corresponding weight% of flame retardants. The additives included increasing concentrations of DMMP, TEP, or EG flame retardants with 0.18 g of A-1, 0.04 g of T-12, 0.4 g of B8404 surfactant, and 0.8 g of water. First, in component A, the synthesized HSPO, commercial polyol SG-522 (50/50 wt./wt. ratio), and the additives were mixed in a disposable plastic cup using a high-speed mechanical stirrer for 1–2 min to form a uniform mixture. Then, the weighted part component B (isocyanate) was added to the blended component A, and the mixture was stirred at 2000–2500 rpm for 20–30 s. The mixture was poured into the mold, where the foam started to rise freely. Afterward, the foam was cured at room temperature for 7 days to complete the polymerization. The effect of FRs on the intrinsic flame retardancy of the foams was studied. The RPUFs with 0 to 10.61 wt% of all the FRs were prepared similarly. Increasing the flame retardant concentration above 10% leads to non-uniform dispersion of FRs in the polymer matrix. The higher addition of the flame retardants also increases the viscosity of the polyol premix which makes it difficult to uniformly mix with the isocyanate, thereby limiting the foam growth.
Characterization of EHSO and HSPO
The synthesized epoxidized hempseed oil and the polyol were characterized through several standardized analytical techniques. The iodine content of the hemp seed oil was established using the Hanus method. The hydroxyl number of the HSPO was determined using phthalic anhydride pyridine (PAP), following ASTM-D4274. Additionally, the percentage of oxirane oxygen (EOC %) of the HSPO was assessed using glacial acetic acid and tetraethylammonium bromide. PerkinElmer Spectrum Two FT-IR spectrophotometer was used to confirm the formation of EHSO and HSPO from HSO. The molecular weights of the HSO, EHSO, and HSPO samples were evaluated using Waters (Milford, MA, USA) gel permeation chromatography (GPC) instrument at 30 °C equipped with 5 μm phenogel columns and tetrahydrofuran (THF) as the eluent solvent (flow rate of 1 mL/min). The viscosity of the samples was measured using a TA Instruments AR 2000 dynamic stress rheometer (Delaware, USA) fitted with a plate angle of 2° and a cone plate diameter of 25 mm.
Characterization of HSO-Based Rigid Polyurethane Foams
The HSO polyurethane foams were cut into uniform sizes using a sharp knife to determine the thermal and physio-mechanical properties. The apparent density of the rigid foam samples was measured according to the ASTM D1622 method as a ratio of the masses and volumes of specimens. The core cell morphology of the foams was characterized by a scanning electron microscope (SEM) (Phenom, The Netherlands). SEM was performed with a constant acceleration voltage of 10 kV in a chamber under a high vacuum. Foam cube specimens of 0.5 cm3 were cut and gold surface sputtering was used for analysis.
The foams’ closed cell content (CCC) was evaluated using an Ultrapycnometer, Ultrafoam 1000 following the ASTM 2856 standard method. The compressive strength of the HSO-RPUFs at 10% deformation was measured on a universal testing machine (MTS, USA) along the direction perpendicular to the foam growth, following the ASTM 1621 method. Sample dimensions of 50 × 50 × 25 mm3 (L) × (B) × (H) were cut from the cores of polyurethane foams and a strain rate of 30 mm/min was applied for analysis.
The thermal decomposition behavior of the foams was analyzed using the Q500 TA instruments (Delaware, USA) thermogravimetric analyzer under the N2 atmosphere. 8–10 milligrams of foam sample were placed in the aluminum crucible and heated from 30 to 700 °C with a ramp rate of 10 °C/min. A flammability test was conducted according to ASTM D4986-18 standard test procedure to examine the effects of DMMP, TEP, and EG on the fire retardancy of the HSO-based foams. The foams of specific dimensions 150 (height) × 50 (length) × 12.5 (thickness) mm3 were placed horizontally and exposed to flame applied perpendicularly for 10 s. The total time for the sample to extinguish the fire was noted during the burning process. The specimen’s pre- and post-burning weights were recorded to calculate the weight loss percentage.
Results and Discussion
Characterization of Epoxidized HSO and HSO-Polyol
The iodine value of the HSO was evaluated as 124.31 g of I2/100 g. The HSO-epoxide was characterized by the epoxy content, EOC % as 5.9 mol/100 g of oil. The synthetic hempseed polyol demonstrated an acid value of 0.56 mg KOH/g and a hydroxyl number of 209 mg KOH/g. FT-IR, GPC, viscosity, and other supporting tests were carried out to evaluate and confirm the entire synthesis of the HSO-polyol.
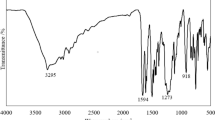
The formation of the epoxidized oil and polyols was confirmed via FT-IR spectra, as shown in Fig. 2a. In the FT-IR spectra of the HSO, the peak at 3005 cm-1 is attributed to the = C-H groups stretching vibration present, which is also indicative of unsaturation in vegetable oils [33]. The disappearance of = C-H and C = C stretching peaks and the appearance of oxirane ring vibration peak (C-O-C bending) at 839 cm-1 in the EHSO, confirmed the transformation of the epoxidized HSO from the virgin HSO. In the case of HSO-polyol, the emergence of a broad hydroxyl (-OH) peak around 3462 cm-1 and the fading away of the epoxy vibration peak at 839 cm-1 suggests the formation of the polyol after successful ring opening with methanol. Furthermore, the HSO-polyol showed a higher viscosity of 2.54 Pa.s in comparison to EHSO and HSO, obtained as 0.07 Pa.s and 0.15 Pa.s respectively. The higher viscosity of the HSO-polyols is a sign of the increase in molecular weight following the conversion to polyol from initial oils.
Figure 2b shows the GPC chromatograms of HSO, EHSO, and HSPO. The peaks with a 32.3 min retention time are the result of hempseed triglyceride oil. The chromatograms of EHSO showed a retention time of 32.7 min. In the case of HSPO, there was also a shoulder peak showing the formation of dimers with a retention time of 30.7 min and a peak demonstrating increased molecular weight of polyol with a retention time of 31.8 min. During the epoxide ring-opening reaction, numerous side reactions take place, including oligomerization reactions [34]. The appearance of peaks at lower retention times comes from the formation of dimers, trimers, and oligomers, respectively [34, 35].

(a) FTIR and (b) GPC analysis of the HSO, EHSO, and HSPO
Density and Closed Cell Content of HSO-RPUFs
The surface and mechanical properties of the resulting RPUFs are significantly influenced by their density. The properties of the polyurethane foams are listed in Table 2. According to the results, the density of RPUFs containing different flame retardants (DMMP, TEP, and EG) was found lower than that of neat RPUFs. With increasing concentrations of various flame retardants, the foam densities were obtained between 37 and 47 kg/m3. The densities of most industrially applicable commercial rigid foams fall between 20 and 50 kg/m3, which is comparable to the fabricated HSO-RPUFs [36]. The average density of the DMMP-based RPUFs was between 37 and 42 kg/m3. However, with increasing TEP concentration, the density of the foams steadily rises from 40 kg/m3 to 43 kg/m3. Moreover, it was discovered that adding TEP and DMMP at large loadings had a negligible impact on foam density compared to neat RPUFs. In addition, the densities of EG-infused RPUFs foams were obtained between 40 and 47 kg/m3, with only minor variations adequate for commercial use. The reason for lower densities of FRs-based RPUFs might be the void formation in the foam’s cell structures, which had a substantial impact on the foam’s density. This observation is in accord with earlier reported literature studies [37]. In contrast to DMMP and TEP, the density of RPUFs containing EG was found more at higher concentrations due to the decrease in the average cell size and formation of uniform cell structure, as seen from the SEM analysis.
High closed cell content in a PU rigid foam is necessary for good insulating performance. Table 2 displays the closed-cell content (CCC) of the RPUFs foams with varying FRs loading. All the hempseed oil-based PU foams have comparable closed cell contents (> 90%) to the control foam. At higher concentrations of DMMP and EG, the foam’s CCC slightly increases with no significant variation. Overall, with a closed-cell capacity of more than 90%, all the fabricated foams in our work were shrinkage-free and dimensionally stable. Therefore, it appears that the FRs kept a strong airflow barrier after being added to the foams. The outcomes obtained show great agreement with the current market-available products suited for insulating purposes with no sacrifice to their properties [38].
Compressive Properties of HSO-RPUFs
Numerous parameters, mainly apparent density, cellular size, chemical composition, and filler-polymer interaction affect the overall mechanical properties of polyurethanes [39]. The compression stress-strain properties and compressive strength at 10% deformation of the foams with different FR loadings are presented in Fig. 3; Table 2, respectively. The typical compression curves (stress-strain) of cellular materials like PU foams exhibit three deformation regimes: linear elastic, plateau, and densification. It is noticed that at 10% deformation, neat foams presented a yield strength of 0.210 MPa (210 kPa) i.e., before this point, the foam will follow the linear elastic behavior (stress is proportional to deformation) followed by the irreversible plateau region (plastic deformation). The plateau region is linked with cell collapse [40]. As expected, the compressive strength decreases as the content of different FRs in the polyurethane formulation increases. Following the addition of 10.61 wt% of DMMP (HSO-DMMP-6) and TEP (HSO-TEP-6), the compressive strength decreased to 0.143 and 0.180 MPa, respectively. This observation is probably due to the plasticizing effect of the phosphorous-based FRs on the foam, which leads to the deterioration in mechanical properties [41, 42]. The mechanical properties of the foams are closely correlated with their apparent density. The reduction in the apparent density of the DMMP and TEP-loaded foams reflects the compression characteristics’ decline. Also, the compressive strength values of the DMMP-EG-6 (10.61 wt% EG) were found lower at 0.183 MPa (183 kPa) compared to neat foam. Additionally, as observed from the SEM images specifically, the rise in FRs concentration caused a homogeneous distribution of large and small pores across the fabricated PU and a possible partial breakdown of the cellular structure. These factors reduced the number of cells in the foams; thus, it was anticipated that the compressive strength would decrease as well [40].

Compressive stress-strain curves of the HSO-RPUFs showing linear elastic and plateau regions with different FRs loadings
SEM Morphology of HSO-RPUFs
The influence of different FRs on the morphologies and cellular structure of the PUR foams was examined using scanning electron microscopy (Fig. 4). The average pore size for the control sample was around 310 µm. Irrespective of the type of FRs used, all the foams demonstrated a well-defined closed-cell structure. DMMP-containing foams showed a progressive decrease in the foam cell size as the concentration was raised. The average cell size was calculated around 215–264 µm (Fig. 4a). Similarly, for TEP and EG-based RPUFs, the cell morphology maintained a relatively uniform spherical shape at lower loadings. However, at higher concentrations, the foam cell size increases with non-uniform cellular morphology for both TEP and EG-incorporated RPUFs (Fig. 4b-c). The TEP-based RPUFs had an average cell size ranging from 230–258 µm, whereas the EG-containing foams displayed pore sizes of approximately 256–373 µm. The additional pore size increase may be due to the cells collapsing which is correlated to the mild increase in the density of the EG-containing foams at higher loadings [43, 44]. The above observation suggests that there was a beneficial interaction between the FRs and the HSO-based polyurethane matrix. A frequency distribution plot of the pore-size area (Fig. 4a’-c’) shows the number of large pores relative to small pores in the images. All the imaging systems showed a homogeneous distribution of large and small pores across the fabricated PU foam.

(a-c) SEM images and (a’-c’) frequency distribution of pore-size area of the HSO-RPUFs with different loadings of DMMP, TEP, and EG
Flammability of the HSO-RPUFs
The effect of different FRs on the flame retardancy of the hempseed oil-based rigid foams was tested as per the ASTM D4986-18 standard. The samples were placed on the stand vertically, and a flame was used to ignite them for ten seconds [45]. After the flame was extinguished, the samples’ burning time and weight loss were recorded and are shown in Fig. 5a-b. To confirm the better homogenization and uniform migration of FRs into the foam matrix, the burning test was performed twice by heating the bottom and top of the sample, and the average time was noted. The longer the extinguishing time, the greater the weight loss observed in the specimens. The neat foam (without FRs) had the greatest weight loss, losing almost 81 wt% of its original body weight and burning for about 104 s. The cause could be the irregular and improperly disrupted cells in the neat PU, which contain trapped air and support prolonged burning [46, 47]. DMMP-incorporated foam samples showed a substantial linear decline in the foams’ burning duration and weight reduction. The burning time and the weight loss reduced significantly to 15 s and 6 wt% for 10.61 wt% DMMP (HSO-DMMP-6) foams. A similar trend was observed for TEP and EG-based foams, the burning time and weight loss drastically lowered to 21 s and 4 wt% (HSO-TEP-6), and 13 s and 3 wt% (HSO-EG-6).
Understanding the flame retardancy mechanism is crucial to determine how effective flame retardants are overall at preventing fires. Phosphorus compounds (DMMP and TEP) reduce the production of combustible gases by acting as char-forming agents. It is well reported that DMMP generates unstable free radicals like PO2. and PO. in the gaseous phase and can react exothermically with different byproducts. These decomposed phosphorus radicals produce phosphoric acid which promotes the dehydration reaction useful for reducing heat in the polymer matrix and establishing a char layer to prevent the remaining polyurethane foam from combusting. This makes it harder for flames to start, which lowers quickly when they propagate throughout the foams [46,47,48,49,50].

(a) Burning time and (b) weight loss (%) content of the HSO-RPUFs with varying amounts of DMMP, TEP, and EG
The flame-retardant phenomenon of EG is different from phosphorus compounds and the efficiency of EG is influenced by its size, amount, and density [51]. When exposed to heat, the intercalated graphite particle expands or exfoliates, releasing water vapor, CO2, and SO2 gases and the expanded carbon layer functions as an insulating layer to lessen the heat transfer. Therefore, once exposed to combustion, EG causes a graphitic char layer to grow, forming a worm-like thermal insulating layer structure on the foam surface that effectively blocks the heat and oxygen transfer enhancing fire-retardant performance [51, 52]. The photographs for the horizontal burning test of HSO-based RPUFs with different FRs are shown in Fig. 6a-c. As observed, the anti-flaming properties of RPUFs were greatly improved and enhanced by the addition of different flame retardants in the foam formulation to its neat counterpart. Also, the continuous decrease of self-extinguish burning time of RPUFs with increasing loadings suggests better homogenization and uniform migration of FRs into the foam matrix.

Picture of HSO-RPUFs from the horizontal burning test: (a) DMMP, (b) TEP, and c) EG
Thermogravimetric Analysis of the HSO-RPUFs
To further understand the thermal stability of the neat and FRs containing HSO-RPUFs, TGA and derivative thermogravimetric analysis (DTGA) were conducted. Figure 7a-f indicates the TGA curves of the foam samples as a function of temperature and weight loss under investigation. The neat PU sample (without any flame retardant) exhibits two-step degradation stages. The first step between 200 and 350 °C is due to degradation of the hard segments i.e., breaking of the urethane linkage [46, 53]. The second step is associated with the soft segments’ thermal breakdown, observed from 350 to 450 °C. However, DMMP and TEP-incorporated foams registered a three-step degradation pattern (Fig. 7a and c). The first decomposition peak (80–200 °C) as observed was thought to be due to the partial vaporization and breakdown of DMMP and TEP FRs, which will facilitate the formation of a condensed char later in the thermal degradation process [54, 55]. The second weight loss i.e., from 200 to 350 °C is attributed to the degradation of the urethane linkages followed by polyol decomposition (> 400 °C). All the DMMP and TEP-based foams with varying concentrations present essentially similar degradation profiles. Notably, foams with higher DMMP or TEP concentrations showed less weight loss than neat foam, indicating improved thermal stability. Foams with a higher concentration of DMMP have higher residual char yield (more than 12 wt%) whereas the clean foam yields 8 wt%. char residue.
On the other hand, PU foams incorporated with EG exhibited a two-step thermal degradation mechanism, as displayed in Fig. 7e and f. In this view, the initial breakdown was observed from 250 to 400 °C, which is attributable to the thermal cleavage of the hard portions, and urethane linkage, as previously mentioned. The second decomposition was connected to the subsequent breakdown of isocyanate and polyol i.e., soft segments [46, 53]. Moreover, the irreversible exfoliation of EG results in the emission of H2O and CO2, SO2 gases between 250 and 300 °C temperature. Also, the foams with increasing concentrations of EG presented a higher residue (> 20 wt%.) than the neat foam, perhaps due to the development of the worm-like carbonaceous insulating char layer that slows down the material from complete degrading [56]. These findings illustrate the overall improvement of foams’ thermal stability following the incorporation of DMMP, TEP, and EG-based FRs into the polyurethane formulation. Additionally, the TGA results were consistent with those of the residual weight loss percentage obtained from the flammability tests, indicating an improvement in overall thermal stability after the addition of FRs.

Thermal analysis for HSO-RPUFs with varying amounts of FRs
Conclusions
In summary, we demonstrate a successful synthesis of bio-based rigid polyurethane from hemp seed oil for the first time. The bio-polyol was produced by formal epoxidation followed by ring-opening of crude hempseed oil with peracetic acid generated in situ by the reaction of acetic acid and hydrogen peroxide. The polyurethane foams produced with different FRs like DMMP, TEP, and EG did not differ significantly in terms of their apparent density, closed cell content, and compressive strength properties. It is noteworthy that the addition of FRs to the polyurethane matrix led to some variance in the foam morphology supported by the increase in cell size and some structural imperfections. However, even with the high loadings of FRs in every formulation, no cell damage was noticed. The effect on compressive strength variation was not significant, where the HSO-DMMP-6, HSO-TEP-6, and HSO-EG-6 RPUFs displayed compressive strength of 143, 180, and 183 kPa compared to neat foam yield strength (10% deformation) of 210 kPa. The inclusion of different non-halogenated FRs resulted in a notable improvement in the thermal stability of RPUFs, as observed from TGA. Finally, the burning test revealed that loading of 10.6 wt% of DMMP, TEP, and EG individually presented lower weight loss and lesser burning times of 15 s and 6 wt%, 21 s, and 4 wt% and 13 s and 3 wt%, respectively compared to the control sample (104 s and 81 wt%). This suggests that DMMP and EG-incorporated foams exhibited better flame-retarding properties as compared with TEP. The resulting biobased hempseed oil-based rigid polyurethane foams combined with various FRs are anticipated to be highly appropriate for the next generation of lightweight, anti-flammable materials used in various engineering and industrial applications, particularly in the building and construction materials sector, due to their enhanced mechanical, thermal, and anti-flammable qualities.
Further research is required, especially limiting oxygen index (LOI), and cone calorimetry tests to overcome the shortcomings of flame retardant efficiency and migration of FRs in the PU matrix, for better reach. Also, the synergetic effect between phosphorus and expandable graphite-based flame retardants in RPUFs for improved fire-safety performances can be explored in the scope of future studies.
References
Szycher M (1999) Handbook of polyurethanes. CRC, Boca Raton, FL
Gama NV, Ferreira A, Barros-Timmons A (2018) Polyurethane foams: past, present, and future. Materials 11:1841
Akindoyo JO, Beg MDH, Ghazali S, Islam MR, Jeyaratnam N, Yuvaraj AR (2016) Polyurethane types, synthesis and applications-a review. RSC Adv 6:114453–114482
Wendels S, Av´erous L (2021) Biobased polyurethanes for biomedical applications. Bioact Mater 6:1083–1106
Engels HW, Pirkl HG, Albers R, Albach RW, Krause J, Hoffmann A, Casselmann H, Dormish J (2013) Polyurethanes: versatile materials and sustainable problem solvers for today’s challenges. Angew Chem Int Ed 52:9422–9441
Hai TAP, Tessman M, Neelakantan N, Samoylov AA, Ito Y, Rajput BS, Pourahmady N, Burkart MD (2021) Renewable polyurethanes from sustainable Biological precursors. Biomacromolecules 22:1770–1794
Zhu Y, Romain C, Williams C (2016) Sustainable polymers from renewable resources. Nature 540:354–362
Xia Y, Larock RC (2010) Vegetable oil-based polymeric materials: synthesis, properties, and applications. Green Chem 12:1893–1909
Sardon H, Mecerreyes D, Basterretxea A, Avérous L, Jehanno C (2021) From lab to market: current strategies for the production of Biobased Polyols. ACS Sustain Chem Eng 9:10664–10677
Li Y, Luo X, Hu S (2015) Polyols and polyurethanes from Vegetable oils and their derivatives Á vegetable oils Á fatty acids Á. Springer, Berlin/Heidelberg, Germany
Cabulis U, Ivdre A (2023) Recent developments in the sustainability of the production of polyurethane foams from polyols based on the first- to the fourth-generation of biomass feedstock, current opinion in Green and Sustainable Chemistry, 44, 100866
Kirpluks M, Kalnbunde D, Benes H, Cabulis U (2018) Natural oil-based highly functional polyols as feedstock for rigid polyurethane foam thermal insulation. Ind Crops Prod 122:627–636
Petrovic ZS (2008) Polyurethanes from Vegetable oils. Polym Rev 48:109–155
Zhang C, Garrison TF, Madbouly SA, Kessler MR (2017) Recent advances in Vegetable Oil-based polymers and their composites. Prog Polym Sci 71:91–143
Miao SD, Zhang SP, Su ZG, Wang P (2013) Synthesis of Biobased polyurethanes from Epoxidized Soybean Oil and Isopropanolamine. J Appl Polym Sci 127:1929–1936
Lu YS, Larock RC (2008) Soybean-oil-based Waterborne polyurethane dispersions: effects of Polyol Functionality and Hard Segment Content on properties. Biomacromolecules 9:3332–3340
Bähra M, Mülhaupta R (2012) Linseed and soybean oil-based polyurethanes prepared via the non-isocyanate route and catalytic carbon dioxide conversion. Green Chem 14:483–489
Zhang CQ, Xia Y, Chen RQ, Huh S, Johnston PA, Kessler MR (2013) Soy-Castor Oil-based polyols prepared using a Solvent free and Catalyst-free Method and polyurethanes Therefrom. Green Chem 15:1477–1484
Calvo-Correas T, Mosiewicki MA, Corcuera MA, Eceiza A, Aranguren MI (2015) Linseed oil-based polyurethane rigid foams: synthesis and characterization. J Renew Mat 3:3–13
Tanaka R, Hirose S, Hatakeyama H (2008) Preparation and characterization of polyurethane foams using a Palm Oil-based Polyol. Bioresour Technol 99:3810–3816
Zhang C, Madbouly SA, Kessler MR (2015) Biobased polyurethanes prepared from different vegetable oils. ACS Appl Mater Interfaces 7:1226–1233
Bhoyate S, Ionescu M, Kahol PK, Gupta RK (2019) Castor-oil-derived nonhalogenated reactive flame-retardant-based polyurethane foams with significant reduced heat release rate. J Appl Polym Sci 136:47276
Dworakowska S, Bogdal D, Prociak A (2012) Microwave-assisted synthesis of polyols from Rapeseed Oil and properties of Flexible polyurethane foams. Polymers-Basel 4:1462–1477
Burton RA, Andres M, Cole M (2022) Industrial Hemp seed: from the field to value-added food ingredients. J Cannabis Res 4:45
Cherney JH, Small E (2016) Industrial Hemp in North America: production, politics and potential. Agronomy 6:58
Kostić MD, Joković NM, Stamenković OS, Rajković KM, Milić PS, Veljković VB (2013) Optimization of hempseed oil extraction by n-hexane. Ind Crops Prod 48:133–143
Li R, Zhang P, Liu T, Muhunthan B, Xin J, Zhang J (2018) Use of Hempseed-Oil-Derived Polyacid and Rosin-Derived Anhydride Acid as Cocuring agents for Epoxy materials. ACS Sustain Chem Eng 6:4016–4025
Moser BR, Cermak SC, Doll KM, Kenar JA, Sharma BK (2022) A review of fatty Epoxide Ring opening reactions: Chemistry, recent advances, and applications. J Am Oil Chem Soc 99:801–842
Manthey NW, Cardona F, Francucci G, Aravinthan T (2013) Thermo-Mechanical properties of Epoxidized Hemp Oil-based bioresins and biocomposites. J Reinf Plast Compos 32:1444–1456
Ho YH, Parthiban A, Thian MC, Ban ZH (2022) Siwayanan, P. Acrylated Biopolymers Derived via Epoxidation and Subsequent Acrylation of Vegetable Oils. Int J Polym Sci 2022: 1–12
Surender R, Mahendran AR, Wuzella G, Vijayakumar CT (2016) Synthesis, characterization and degradation behavior of Thermoplastic polyurethane from hydroxylated hemp seed oil. J Therm Anal Calorim 123:525–533
Benaniba MT, Belhaneche-Bensemra N, Gelbard G (2008) Epoxidation of sunflower oil with peroxoacetic acid in the presence of ion exchange resin by various processes. Energy Educ Sci Technol 21:71–82
Lerma-García MJ, Ramis-Ramos G, Herrero-Martínez JM, Simó-Alfonso EF (2010) Authentication of extra virgin olive oils by Fourier-transform infrared spectroscopy. Food Chem 118:78–83
Guo Y, Hardesty JH, Mannari VM, Massingill JL (2007) Hydrolysis of Epoxidized Soybean Oil in the Presence of Phosphoric Acid. J Am Oil Chem Soc 84:929–935
Gupta RK, Ionescu M, Wan X, Radojcic D, Petrovic ZS (2015) Synthesis of a Novel Limonene based Mannich Polyol for rigid polyurethane foams. J Polym Environ 23:261–268
Lee ST, Ramesh NS (2004) Polymeric foams: mechanisms and materials, vol 1, 1st edn. CRC, Boca Raton, FL, USA, p 360
Thirumal M, Khastgir D, Nando GB, Naik YP, Singha NK (2010) Halogen-free flame retardant PUF: effect of melamine compounds on mechanical, thermal, and flame retardant properties. Polym Degrad Stab 95:1138–1145
Kairytė A, Kremensas A, Balčiūnas G, Członka S, Strąkowska A (2020) Closed cell rigid polyurethane foams based on low functionality polyols: research of Dimensional Stability and standardized Performance Properties. Materials 13:1438
Czupry ´nski B, Paciorek-Sadowska J, Liszkowska J (2010) Properties of rigid polyurethane-polyisocyanurate foams modified with the selected fillers. J Appl Polym Sci 115:2460–2469
Polaczek K, Kura ´nska M (2022) Hemp seed oil and oilseed Radish Oil as New sources of raw materials for the synthesis of Bio-polyols for Open-Cell polyurethane foams. Materials 15:8891
Bhoyate S, Ionescu M, Kahol PK, Chen J, Mishra SR, Gupta RK (2018) Highly flame-retardant polyurethane foam based on reactive phosphorus polyol and limonene-based polyol. J Appl Polym Sci 135:16–19
Asare MA, Kote P, Chaudhary S, de Souza FM, Gupta RK (2022) Sunflower Oil as a renewable resource for polyurethane foams: effects of Flame-Retardants. Polymers 14:5282
Kaur R, Kumar M (2020) Addition of anti-flaming agents in castor oil based rigid polyurethane foams: studies on mechanical and flammable behavior. Mater Res Express 7:015333
Choe H, Choi Y, Kim JH (2019) Threshold cell diameter for high thermal insulation of water-blown rigid polyurethane foams. J Ind Eng Chem 73:344–350
Wang Y, Zhang J (2013) Thermal stabilities of drops of burning thermoplastics under the UL 94 vertical test conditions. J Hazard Matter 246–247:103–109
Chattopadhyay DK, Webster DC (2009) Thermal stability and flame retardancy of polyurethanes. Prog Polym Sci 34:1068–1133
Günther M, Lorenzetti A, Schartel B (2018) Fire phenomena of rigid polyurethane foams. Polymers 10:1166
Camino G, Costa L, di Cortemiglia MPL (1991) Overview of fire-retardant mechanisms. Polym Degrad Stab 33:131–154
Günther M, Levchik SV, Schartel B (2020) Bubbles, and collapses: Fire Phenomena of Flame-Retarded Flexible polyurethane foams. Polym Adv Technol 31:2185–2198
Kirpluks M, Cabulis U, Zeltins V, Stiebra L, Avots A (2014) Rigid polyurethane Foam Thermal Insulation protected with Mineral Intumescent Mat. Autex Res J 14(4):259–269
Bian XC, Tang JH, Li ZM, Lu ZY, Lu A (2007) Dependence of Flame-Retardant properties on Density of Expandable Graphite filled rigid polyurethane foam. J Appl Polym Sci 104:3347–3355
Ming G, Chen S, Sun YJ, Wang YX (2017) Flame retardancy and Thermal properties of Flexible polyurethane foam containing expanded Graphite. Combust Sci Technol 189:793–805
Petrovi´c ZS, Zavargo Z, Flyn JH, Macknight WJ (1994) Thermal degradation of segmented polyurethanes. J Appl Polym Sci 51:1087–1095
Yang L, Shroll RM, Zhang J, Lourderaj U, Hase WL (2009) Theoretical investigation of mechanisms for the gas-phase unimolecular decomposition of DMMP. J Phys Chem A 113:13762–13771
Schartel B (2010) Phosphorus-based flame retardancy mechanisms-old hat or a starting point for Future Development? Materials 3:4710–4745
Lorenzetti A, Dittrich B, Schartel B, Roso M, Modesti M (2017) Expandable Graphite in polyurethane foams: the effect of expansion volume and intercalants on Flame Retardancy. J Appl Polym Sci 134:45173
Acknowledgements
The authors are grateful to the National Institute of Standards and Technology (NIST award number 70NANB20D146) and the U.S. Economic Development Administration (US-EDA award number 05-79-06038) for providing research infrastructure funding.
Author information
Authors and Affiliations
Contributions
S.J.: Investigation, Methodology, Data curation. Y.N.D.: Data curation, Formal analysis. P.S.: Formal analysis, Writing-original draft; Writing-review & editing. R.K.G.: Conceptualization, Resources, Project administration, Funding acquisition, Validation, Writing-review & editing.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing financial interests towards any individual or organization.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jariwala, S., Desai, Y.N., Sahu, P. et al. Hemp Seed Oil Derived Rigid Polyurethane Foams and Their Underlying Flame Retardancy Properties. J Polym Environ 32, 3822–3834 (2024). https://doi.org/10.1007/s10924-024-03215-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-024-03215-7