
Abstract
In this research, bio-based polyamide (bio-PA) was synthesized from dimethyl furan-2,5-dicarboxylate and 1,3-cyclohexanedimethanamine by melt polymerization. The properties of bio-PA were analyzed by Fourier transform infrared spectrometer (FTIR), nuclear magnetic resonance (NMR), differential scanning calorimetry (DSC), X-ray diffraction (XRD), and thermal gravimetric analysis (TGA), respectively. The results show that this bio-PA presents high glass transition temperature (Tg) from 150 °C to 180 °C and poor crystallization due to the asymmetric rigid structure of cyclohexane and furan. Its molecular weight is low, ascribing to the large steric hindrance from cyclohexane and furan, and the side reaction of N-methylation and decarboxylation. Besides, the results of solubility reveal that this bio-PA can be dissolved in DMSO, DMF and DMAC.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyamide (PA) has good comprehensive properties, including mechanical properties, heat resistance, wear resistance, chemical resistance and easy processing, is widely used as one of engineering plastics. It is usually synthesized by polycondensation of diamine and diacid [1], diamine and diacid derivatives or ring-opening polymerization of lactam [2, 3]. At present, the monomer of polyamide, such as terephthalic acid, isophthalic acid, hexamethylene diamine and butane diamine, is mainly derived from petroleum resources, which are non-renewable and cause energy crisis. Nowadays, utilization of biomass to develop bio-based polyamide attracts many researchers attention and becomes a new hotspots [4].
Furandicarboxylic acid (FDCA) is a kind of bio-based commercial compound, is usually generated from 5-(hydroxymethyl)furfural (5-HMF) by chemical [5,6,7,8] or biological method [9]. HMF is mainly derived from natural hexose (glucose, monosaccharide) [10]. The carbon content of FDCA is same as hexose and there is no carbon release during the conversion. Therefore, FDCA has been regarded as one of the 12 most valuable bio-based platform compounds by US Department of Energy, can be used to synthesize the furanyl polyester and polyamide. Compared FDCA with terephthalic acid (TPA), their physicochemical properties are similar. The carbon atom of furan is less than that of benzene and its aromaticity is weak, the bond angle is small and the rigidity is strong. Therefore, the FDCA has been considered as replacement for the fossil based terephthalic acid. At present, the research on furandicarboxylic acid has been greatly developed. For example, FDCA has been applied to synthesize the semicrystalline aromatic polyesters, poly(ethylene 2,5-furandicarboxylate) (PEF) [11] and poly(butylene 2,5-furandicarboxylate) (PBF) [12, 13]. The results show that polyesters based on FDCA exhibits lower Tm and higher Tg than that of polyesters from TPA, which makes it a potential substitute for petroleum-based polyethylene terephthalate (PET) and polybutylene terephthalate (PBT) [14]. Correspondingly, the FDCA-based polyamides as a substitute of traditional aromatic polyamides also attract widespread attention.
As early as 1961, Hopff and Krieger [15] firstly reported the synthesis of bio-based polyamide with furan ring, which broke the dependence of polyamide industry on petroleum resources. Afterwards, Fehrenbacher [16] synthesized poly(hexamethylene furanamide) (PA6F), poly(octamethylene furanamide) (PA8F), poly(decamethylene furanamide) (PA10F), and poly(dodecamethylene furanamide) (PA12F) via melt polycondensation using organometallic catalysts. Yohana [17] synthesized copolyamide of poly(butylene-2,5-furan dicarboxylamide) (PA4F) and poly(butylene adipamide) (PA46) through consecutive prepolymerization and solid-state polymerization. Recently, transparent PA attracted many researchers attention and exhibited potential application in many fields, such as machine parts, optical instruments and sports equipment. However, there are few literatures on the preparation of transparent PA, especially transparent bio-based PA. Transparency is closely associated with crystallinity. Therefore, it is important to develop bio-based amorphous polyamide. Thibault [18] synthesized a series of polyamides and copolyamides by melt polycondensation from FDCA, isophthalic acid (IPA) and 1,6-hexanediamine. It is observed that the polyamides synthesized from dicarboxylic acid or its derivatives present low crystallinity or amorphous structure, the copolyamides exhibit low melting point and crystallinity with the increase of FDCA content ecrea [19,20,21,22]. Wilsens [23] investigated the depression effect of furan rings on the formation of hydrogen bonds among amide groups using small molecule compounds. It found that the oxygen heteroatom in FDCA ring acts as a hydrogen bond acceptor to form intramolecular hydrogen bonds, which reduce the formation of intermolecular hydrogen bonds and interaction forces. This is benefit to prepare the amorphous and transparent PA materials. Besides, 1,3-cyclohexanedimethanamine is a mixture of cis–trans isomer, which destroys the regular structure of polymers and guarantees the high transparency. Its cyclohexane structure is benefit to the high glass-transition temperature.
In this research, bio-based amorphous polyamide was designed and synthesized by melt polymerization from dimethyl furan-2,5-dicarboxylate and 1,3-cyclohexanedimethanamine. The effect of reaction temperature and time on the properties of target product was fully investigated.
Experimental Section
Materials
Dimethyl furan-2,5-dicarboxylate (DMFDCA, 98%), 1,3-cyclohexanedimethanamine (Isomer mixture, CHDM, 99%) was purchased from Tokyo Chemical Industry. Dimethyl sulfoxide-d6 (DMSO) was purchased from Shanghai Yien Chemical Technology Co. Ltd. Sulfuric acid (H2SO4, 98%) was purchased from Pinghu Chemical Reagent Factory.
Synthesis of Polyamide
5.0 g (27.1 mmol) DMFDCA and 3.86 g (27.1 mmol) 1,3-cyclohexanedimethanamine were transferred to a glass tube. Before polymerization (pre-polymerization), the mixture was gradually raised to 120 °C, maintaining for 30 min under the protection of nitrogen. The viscosity of the mixture increases, and a prepolymer was obtained. Then (post- polymerization), the prepolymer was further heated to 230 °C, 240 °C, 250 °C and reacted for different time under vacuum. The pressure was controlled below 300 Pa. The chemical equation is shown in Scheme 1, and the synthesis condition of different PA samples is shown in Table 1. Finally, pale yellow transparent polyamide PAFCHDM was obtained, as shown in Fig. 1.

The equation for synthesis of furan polyamide in this research. DMFDCA refers to dimethyl furan-2,5-dicarboxylate and CHDM refers to 1,3-cyclohexanedimethanamine

Images of the PA synthesized under different conditions
Measurements
For infrared spectra testing, the polymers were ground into powder and carried out on Fourier transform infrared spectrometer (FT-IR) with attenuated total reflectance (ATR) method.
1H and 13C NMR spectra were recorded on a Bruker 600 MHz NMR spectrometer at room temperature. All the compounds were dissolved in dimethyl sulfoxide-d6 (DMSO-d6). The spectra were internally referenced to tetramethylsilane (TMS). About 10 mg and 50 mg sample were dissolved in 5 mL solvent using for 1H NMR and 13C NMR testing, respectively. 108 scans were recorded for 1H NMR and 1280 scans for 13C NMR.
The thermal behavior of polymers was examined by differential scanning calorimetry (DSC). The thermograms were obtained from 5 to 10 mg samples at heating and cooling rates of 20 °C/min from 30 °C to 250 °C under a nitrogen flow of 20 mL/min.
Thermogravimetric analysis (TGA) was performed on TGA 209F1 from 50 °C to 600 °C under nitrogen flow of 20 mL/min at a heating rate of 10 °C/min.
XRD patterns were recorded on Rigaku D/max-2550 using Cu Kα radiation in the scan range from 5° to 90° with increment of 0.02.
For intrinsic viscosity measurement, the samples were dissolved in 98% sulfuric acid to get solutions of 0.5 g/dL and measured with JWC-32C viscometer in 25 ± 0.02 °C water bath. The flow time of solution and sulfuric acid were recorded as t and t0, respectively. The relative viscosity (ηr) and specific diversity (ηsp) were calculated as follows:
The solution concentration was marked as C. The intrinsic viscosity [η] can be calculated by one-point method (Solomon and Ciuta relationship) [24] as follows:
Finally, the viscosity-average molecular weight (Mν) was calculated according to the following equation [25]:
As comparison, Weight-average Molecular Weight (Mw) was measured by Shimadzu Gel Permeation Chromatography CTO-20A (GPC-20A). All the PA samples were dissolved in DMF and polystyrene was used as standard.
Results and Discussion
Structure Analysis of Furan Polyamide
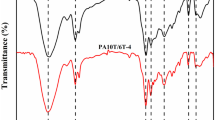
Figure 2 presents the FT-IR spectra of synthesized furan polyamides. The C = C stretching vibrations (1560-1580 cm−1), C–O–C groups (1011 cm−1), and the out-of-plane deformed vibrations of C-H groups (967 cm−1, 820 cm−1, and 755 cm−1), can be found in all the PAFCHDM samples, demonstrating the introduction of furan rings in these polymers. The peak at 1720 cm−1 was observed for the 120–230-60 min sample, which corresponds to the free C = O from COOCH3 and demonstrates the DMFDCA was not fully reacted at 230 °C. When the temperature increases to 240 °C, the peak disappeared, indicating the full conversion of ester to amide groups. The two peaks at 3430 cm−1 and 3294 cm−1 are attributed to the stretching vibrations of “free” and hydrogen bonded N–H in amide groups, suggesting the presence of hydrogen bonds in these polyamides. The formation of amide groups is also confirmed by the presence of amide I, II and III complex vibration bands, which is located around 1652 cm−1, 1530 cm−1 and 1292 cm−1, respectively.

FT-IR spectra of PAFCHDM. (a) stretching vibration of free N–H; (b) stretching vibrations of hydrogen-bonded N–H groups; (c) C-H of furan stretching; (d) C = C stretching vibrations
Figures 3, 4 presents the possible reaction and NMR results of products, respectively. As shown in Fig. 4a, the proton signals at 7.1 ppm is assigned to c and c' of furan rings. The resonances at 8.5 ppm attribute to the proton k of amide bonds. The proton signal peaks at 6.6 ppm and 7.8 ppm are presumed to the hydrogen on c'' and b''. The signal peak at 2.25 ppm may be ascribed to the proton hydrogen of i after N-methylation reaction. From this result, it can be inferred that the N-methylation and decarboxylation occur during the polymerization. This leads to the low molecular weight of PAFCHDM and will be further explained by 13C NMR spectrum.

N-methylation and decarboxylation side reaction during the melt polymerization from DMFDCA and CHDM

a 1H NMR of the synthesized PAFCHDM, including three possible byproducts; b 13C NMR of PAFCHDM, which was dissolved in DMSO-d6; c DEPT spectrum of the PAFCHDM, DEPT spectroscopy is a type of carbon spectroscopy, is used to distinguish the primary carbon, secondary carbon, tertiary carbon and quaternary carbon
Figure 4b, c exhibits the 13C NMR and DEPT spectra of PAFCHDM. DEPT spectroscopy is a type of carbon spectroscopy, is mainly used to distinguish the primary carbon (CH3), secondary carbon (CH2), tertiary carbon (CH) and quaternary carbon (C). The peaks of CH and CH3 are upward, which of CH2 is downward, and no peak appears for the quaternary carbon in DEPT spectrum. In this research, the resonance peaks d, dʹ, dʺ are methylene signals, and the dʹ, dʺ are ascribed to the N-methylation reaction between DMFDCA and CHDM [26, 27]. The resonance peaks b, bʹ, bʺ and c, cʹ, cʺ are attributed to the carbon atom on furan ring. The peaks b', b'' and c', c'' are methine signals derived from the byproduct of decarboxylation. Peaks i and j are attributed to the signal of -CH3 after the single and double substitution of NH2. The N-methylation and decarboxylation blocks the terminal amino group and the steric hindrance increases, leads to the termination of reaction or the reduction of reaction activity, resulting in the low molecular weight of PAFCHDM.
Thermal Properties and Crystallization
The bio-PA PAFCHDM was synthesized according to Scheme 1, their molecular weight was measured by one-point method [24] and all the results are listed in Table 2. It shows that the Mv of PAFCHDM under different conditions is low and almost the same, hasn't obvious change with the temperature and time. That is because 1,3-cyclohexane dimethyl-amine contains a rigid cyclohexane, which is a mixture of cis- and trans-isomers. This leads to the high steric hindrance and makes the movement of diamines difficult. At the same time, Wilsens [28] demonstrated that FDCA requires more energy to become activity than regular phenyl based monomers. DMFDCA is a derivative of FDCA, is rigid and hard to move. This cause the reaction activity between DMFDCA and 1,3-cyclohexanedimethanamine is poor, resulting in the low molecular weight. Most important, N-methylation and decarboxylation is another reason for the low molecular weight of PAFCHDM, as shown in Fig. 3.
Thermal properties and crystallization of the PAFCHDM were analyzed by DSC, XRD and TGA respectively, all the data are summarized in Table 2. Figure 5 presents the DSC and XRD results of selected PAFCHDM samples. As shown in Fig. 5a, it shows that PAFCHDM exhibits a high Tg of 150 °C–180 °C, which is mainly attributed to the rigid structure of furan ring and 1,3-cyclohexanedimethanamine. The Tg value decreases with temperature and time increase. This is different from the general polymerization process. It is inferred that the oligomer cyclization occurs during the polymerization process [29]. At the beginning, ring-like polyamide is rigid and the movement of molecular chains is difficult, which leads to a high Tg. As the reaction continuous, the ring-like molecular chain becomes long and mobility, which leads to the decrease of Tg. In addition, there are no melting endothermic peaks and crystallization peaks in the second heating curve, which proves the amorphous characterization of PAFCHDM. It is mainly because 1,3-cyclohexanedimethanamine (cis–trans isomer mixture) contains a cyclohexane structure, which includes a ship-type and chair-type conformation, destroys the crystallinity. Moreover, the furan ring inhibits the formation of intermolecular hydrogen bonds and reduces the crystallinity in some extent. Figure 5b shows the XRD spectrum of PAFCHDM under different post polymerization temperature. All samples exhibit a similar profile and present a wide diffraction peak around 19°. This indicates that PAFCHDM is difficulty to crystallize and the crystallinity is low, which is benefit to its transparent.

a: Second heating curves of PAFCHDM from 80 °C to 240 °C under nitrogen; b: XRD profiles of all the PAFCHDM samples
The thermogravimetric traces were recorded at the range of 100–600 °C under nitrogen atmosphere, the results are shown in Fig. 6. It indicates that PAFCHDM starts to decompose around 286–314 °C and possesses a Td-max above 400 °C, confirming the good thermal stability. With the temperature increases, the Td-5% increases and the Td-max has no obvious change. That is because the molecular weight of synthesized PAFCHDM has not significant improvement.

TGA a and DTG b curves of PAFCHDM, which is recorded from 100 °C to 600 °C under nitrogen
Solubility of Furan Polyamide
Application of high-performance polymers is often limited by their poor solubility in organic solvents. Therefore, the solubility of PAFCHDM in several solvents was studied. The results are listed in Table 3. It shows that the PAFCHDM can be dissolved in polar aprotic solvents at room temperature, such as DMSO, DMF, DMAC and NMP. The main reason for its good solubility is ascribed to the low molecular weight and amorphous structure, which makes the solvent easy to spread in molecular chains. Additionally, the inhibitory effect of furan ring reduces the hydrogen bond and intermolecular force, improving the solubility.
Conclusions
The bio-based furan polyamide PAFCHDM was synthesized by melt polymerization from dimethyl furan-2,5-dicarboxylate and 1,3-cyclohexanedimethanamine. 1H NMR and 13C NMR demonstrate the successful synthesis of polyamide and the polymerization process is prone to N-methylation and decarboxylation, which leads to the low molecular weight of synthesized PAFCHDM. The results of DSC and XRD show that the furan ring and asymmetric 1,3-cyclohexanedimethanamine inhibits the crystallization, which has an important effect on the polymer crystallinity. It was also found that the PAFCHDM could be dissolved in DMSO, DMF, DMAC and NMP at room temperature.
References
Christopher SM, Agathe B, Andrew P, Vikas M, John RD (2016) Biorenewable blends of polyamide-4,10 and polyamide-6,10. J Appl Polym Sci 133:43626. https://doi.org/10.1002/app.43626
Winnacker M, Sag J (2018) Sustainable terpene-based polyamides via anionic polymerization of a pinene-derived lactam. Chem Commun 54:841–844. https://doi.org/10.1039/c7cc08266e
Winnacker M, Sag J, Tischner A, Rieger B (2017) Sustainable, stereoregular, and optically active polyamides via cationic polymerization of epsilon-lactams derived from the terpene beta-pinene. Macromol Rapid Commun 38:1600787. https://doi.org/10.1002/marc.201600787
Sousa AF, Vilela C, Fonseca AC, Matos M, Freire CSR, Gruter GJM, Coelhob JFJ, Silvestre AJD (2015) Biobased polyesters and other polymers from 2,5-furandicarboxylic acid: a tribute to furan excellency. Polym Chem 6:5961–5983. https://doi.org/10.1039/c5py00686d
Liu KJ, Zeng TY, Zeng JL, Gong SF, He JY, Lin YW, Tan JX, Cao Z, He WM (2019) Solvent-dependent selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid under neat conditions. Chinese Chem Lett 30:2304–2308. https://doi.org/10.1016/j.cclet.2019.10.031
Chen RR, Xin JY, Yan DX, Dong HX, Lu XM, Zhang SJ (2019) Highly efficient oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid with heteropoly acids and ionic liquids. Chemsuschem 12:2715–2724. https://doi.org/10.1002/cssc.201900651
Chen H, Wang JT, Yao Y, Zhang ZH, Yang ZZ, Li J, Chen KQ, Lu XY, Ouyang PK, Fu J (2019) Cu−Ni bimetallic hydroxide catalyst for efficient electrochemical conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid. ChemElectroChem 6:5797–5801. https://doi.org/10.1002/celc.201901366
Zheng LF, Zhao JQ, Du ZX, Zong BN, Liu HC (2017) Efficient aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on Ru/C catalysts. Sci China Chem 60:950–957. https://doi.org/10.1007/s11426-016-0489-3
Koopman F, Wierckx N, Winde JH, Ruijssenaars HJ (2010) Efficient whole-cell biotransformation of 5-(hydroxymethyl)furfural into FDCA, 2,5-furandicarboxylic acid. Bioresource Technol 101:6291–6296. https://doi.org/10.1016/j.biortech.2010.03.050
Zhou JD, Cao F, Yu ZL, Wen BB, Cui LY, Tang ZQ, Huang T, Wei P (2016) Research progress in preparation and application of bio-based 2,5-furandicarboxylic acid as polyester monomer. Acta Polym Sin 1:1000–3304. https://doi.org/10.11777/j.issn1000-3304.2016.15142
Burgess SK, Leisen JE, Kraftschik BE, Mubarak CR, Kriegel RM, Koros WJ (2014) Chain mobility, thermal, and mechanical properties of poly(ethylene furanoate) compared to poly(ethylene terephthalate). Macromolecules 47:1383–1391. https://doi.org/10.1021/ma5000199
Zhu JH, Cai JL, Xie WC, Chen PH, Gazzano M, Scandola M, Gross RA (2013) Poly(butylene 2,5-furan dicarboxylate), a biobased alternative to PBT: synthesis, physical properties, and crystal structure. Macromolecules 46:796–804. https://doi.org/10.1021/ma3023298
Ma JP, Yu XF, Xu J, Pang Y (2012) Synthesis and crystallinity of poly(butylene 2,5-furandicarboxylate). Polymer 53:4145–4151. https://doi.org/10.1016/j.polymer.2012.07.022
Chi DQ, Liu F, Na HN, Chen J, Hao CC, Zhu J (2018) Poly(neopentyl glycol 2,5-furandicarboxylate): a promising hard segment for the development of bio-based thermoplastic poly(ether-ester) elastomer with high performance. ACS Sustain Chem Eng 6:9893–9902. https://doi.org/10.1021/acssuschemeng.8b01105
Hopff H, Kriege A (1961) Uber polyamide aus heterocyclischen dicarbonsiiuren. Die Makromol Chemie 47:93–113. https://doi.org/10.1002/macp.1961.020470109
Grosshardt M, Fehrenbacher U, Kowollikher K, Tubke B, Dingenouts N, Wilhelm M (2009) Synthese und charakterisierung von polyestern und polyamiden auf der basis von furan-2,5-dicarbonsäure. Chem Ing Tech 81:1829–1835. https://doi.org/10.1002/cite.200900090
Endah YK, Han SH, Kim JH, Kim NK, Kim WN, Lee HS, Lee H (2016) Solid-state polymerization and characterization of a copolyamide based on adipic acid, 1,4-butanediamine, and 2,5-furandicarboxylic acid. J Appl Polym Sci 133:43391. https://doi.org/10.1002/app.43391
Cousin T, Galy J, Rousseau A, Dupuy J (2018) Synthesis and properties of polyamides from 2,5-furandicarboxylic acid. J Appl Polym Sci 135:45901. https://doi.org/10.1002/app.45901
Miyagawa N, Suzuki T, Okano K, Matsumoto T, Nishino T, Mori A (2018) Synthesis of furan dimer-based polyamides with a high melting point. J Polym Sci Polym Chem 56:1516–1519. https://doi.org/10.1002/pola.29031
Jiang Y, Maniar D, Woortman AJJ, Loos K (2016) Enzymatic synthesis of 2,5-furandicarboxylic acid-based semi-aromatic polyamides: enzymatic polymerization kinetics, effect of diamine chain length and thermal properties. RSC Adv 6:67941–67953. https://doi.org/10.1039/c6ra14585j
Jiang Y, Loos K (2016) Enzymatic synthesis of biobased polyesters and polyamides. Polymers 8:243. https://doi.org/10.3390/polym8070243
Jiang Y, Maniar D, Woortman AJJ, Ekenstein GORA, Loos K (2015) Enzymatic polymerization of furan-2,5-dicarboxylic acid-based furanic-aliphatic polyamides as sustainable alternatives to polyphthal-amides. Biomacromol 16:3674–3685. https://doi.org/10.1021/acs.biomac.5b01172
Wilsens CHRM, Deshmukh YS, Noordover BAJ, Rastogi S (2014) Influence of the 2,5-furandicarboxamide moiety on hydrogen bonding in aliphatic-aromatic poly(ester amide)s. Macromolecules 47:6196–6206. https://doi.org/10.1021/ma501310f
Solomon OF, Ciuta IZ (1962) Determination de la viscosite intrinseque de solutions de polymeres par une simple determination de la viscosite. J Appl Polym Sci 6:683–686. https://doi.org/10.1002/app.1962.070062414
Schindler A, Harper D (1979) Viscosity-molecular weight relationships and unperturbed chain dimension. J Polym Sci Polym Chem 17:2593–2599. https://doi.org/10.1002/pol.1979.170170831
Simth DD, Flores J, Aberson R (2015) Polyamide containing the bio-based 2,5-furancarboxylic acid. WO2015059047 A1, United States Patent.
Duursma A, Aberson R, Simth DD (2015) Process for preparing a furan-based polyamide, a furan-based oligomer and compositions and articles comprising the furan-based polyamide. WO2015060718 A1, United States Patent
Wilsens CHRM, Verhoeven JMGA, Noordover BAJ, Hansen MR, Auhl D, Rastogi S (2014) Thermotropic polyesters from 2,5-furandicarboxylic acid and vanillic acid: synthesis, thermal properties, melt behavior, and mechanical performance. Macromolecules 47:3306–3316. https://doi.org/10.1021/ma500433e
Wegkopf K, Meyerhof G (1986) Kinetics of polyamidation in solution, 2. reaction of diphenyl terephthalate and trimethylhexamethylene diamine in dimethylsulfoxide. Die Makromol Chem 187:411–421. https://doi.org/10.1002/macp.1986.021870220
Acknowledgements
The authors greatly acknowledge the financial support from National Key Research and Development of China (Grant Number 2017YFB0309400) and the Fundamental Research Funds for the Central Universities (2232020D-10).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests to influence the work reported in this research.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mao, L., Pan, L., Ma, B. et al. Synthesis and Characterization of Bio-Based Amorphous Polyamide From Dimethyl furan-2,5-dicarboxylate. J Polym Environ 30, 1072–1079 (2022). https://doi.org/10.1007/s10924-021-02265-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-021-02265-5