
Abstract
In this study water soluble sodium carboxymethyl cellulose (CMC) was blended with high density polyethylene (HDPE) by peroxide-initiated melt compounding technique. The compatibility of the blended polymers were carried out by silane crosslinking agent. A series of blends were prepared by varying the CMC contents up to a maximum of 50 phr. The physical properties of non-crosslinked and crosslinked blends were investigated in detail. FTIR analysis of crosslinked blend confirmed the presence of Si–O–Si and Si–O–C absorption peaks at 1050 and 1159 cm−1. Thermal stability of crosslinked blends improved as compared to its non-crosslinked congener. Rheological study of crosslinked blends illustrated high complex viscosity and dynamic shear storage modulus. The tensile strength of virgin polyethylene was 8.1 MPa whereas the maximum tensile strength of 19.6 MPa was observed in crosslinked blend. Similarly lower deformation was observed in crosslinked blends under static load. Scanning electron microscopy of crosslinked formulations also showed strong adhesion between the polymers interface. The compatibility of HDPE and CMC is attributed to both free radical and condensation reactions.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyethylene has many uses in our daily life and its utilization is continuously growing. It is widely used for short term applications such as packaging film, construction, healthcare, agricultural, automobile and are disposed off at dump places as waste. Polyethylene is very resistant to biodegradation and its complete degradation takes several years and the environment suffers long term pollution [1]. Although recycling of polymeric waste is a better solution but it is very costly. Moreover, the recycled products have poor properties but still acceptable for many uses. [2].
Many synthetic biodegradable polymers such as poly-lactic acid [3] and polycaprolactone [4] were developed and commercialized but they were very costly with restricted shelf life. Alternatively, polymer blends and composites containing natural polymers as biodegradable additives (such as chitosan, starch, cellulose and their derivatives) were developed [5, 6]. These composites are easily processed and commercialized. The major disadvantage of bio-polymers addition into synthetic polymers are their compatibility. Most of synthetic polymers are hydrophobic whereas, majority of the natural polymers are hydrophilic in nature except lignin and some other polymers. The resultant blend of two different types of polymers is nearly immiscible. The poor interaction between the blended components causes weaker mechanical propertie [7]. In order to improve the interaction, modifications of both natural and synthetic polymer have been made [8–11].
Sodium carboxymethyl cellulose is an important cellulose derivatives with carboxymethyl groups bound to some of the hydroxyl groups of the glucopyranose ring of the cellulose backbone [12]. CMC has many useful properties, such as water solubility [13], binding ability [14–17], biodegradablity [18], disintegrant [19] and emulsifying agent [20]. It has been used as an important additive to improve the processing of value added products in various industrial sectors such as cosmetics, food stuffs, pharmaceuticals, paper and textile industries [21].
In this article, CMC was blended with HDPE by melt compounding technique. A melt, compatibilization of the blended polymers was carried out by silane crosslinking agent. This work primarily focused on the compatibility and physical properties of HDPE/CMC blends in terms of thermal stability, mechanical properties, melt rheology and morphology. These blends have potential applications in packaging industry.
Experimental
Materials
HDPE with melt flow index of 2.2 g/10 min at 190 °C under 2.16 kg load supplied by Sigma Australia. Carboxymethyl cellulose is commercially available as white granules. Its average molecular weight is 90,000 g/mol and melting temperature is 274 °C. The auto-ignition temperature of CMC is 698 °F. Carboxymethyl cellulose is obtained from Sigma Aldrich, Australia. Vinyl triethoxysilane (VTES) are available in liquid form. The boiling point of VTES is 160 °C. The densities of VTES at 25 °C is 0.903 g/mL. The purity of VTES is 98% and is purchased from Sigma Aldrich, Australia. Dicumyl peroxide (DCP) was used as free radical initiator in this study. The purity of DCP is 98% and its molecular weight is 270.37 g/mol. The melting point and density of DCP are 41 °C and 1.56 g/mL at 25 °C, respectively. It is supplied by Sigma Aldrich, Australia. Technical grade of dibutyltin dilaurate (DBTDL) was used as hydrolysis catalyst. Its function is to facilitate the crosslinking reaction at 90 °C. The molecular weight and density of DBTDL are 631.56 and 1.066 g/ml at 25 °C, respectively. Reagent grade stearic acid having 95% purity was used as lubricant for the better dispersion of filler/additives in the composite. The melting point and boiling point of stearic acid are 72 and 361 °C, respectively. Both DBTDL and stearic acid is provided by Sigma Aldrich, Australia. The chemicals were used without further purification.
Sample Preparation
The CMC was dried in vacuum oven for 24 h at 80 °C. The moisture content of CMC was measured using Sartorius moisture analyzer. The moisture content before and after drying the CMC was 13.7 and 3.1% respectively. A series of HDPE/CMC blends with weight ratio of 100/0, 100/20, 100/30, 100/40, 100/50 were prepared using Brabender internal mixer at 170 °C with rotor speed of 40 rpm (Table 1). The optimum quantities of Stearic acid, DBTDL, DCP and VTES were selected according to our previous study [22]. For crosslinked blends, following steps were taken. In all crosslinked formulations, fixed amounts of dicumyl peroxide (DCP) (0.15 phr, part per hundred parts of resin) and DBTDL (0.05 phr) were dissolved in dry acetone and were sprayed over HDPE pellets. In order to remove acetone the HDPE pellets were dried at 60 °C in oven for 5 min. The DCP coated HDPE pellets were mixed with CMC and stearic acid (0.3 phr) in internal mixer for 3 min at (T = 130 °C, rotor speed = 33 rpm). Fixed amount of VTES (2.8 phr) was added for 2 min to the mixture during melting stage. For the next 7 min, the temperature and the rotor speed were fixed at 170 °C and 40 rpm respectively. Finally, the blended material was heat pressed into sheets at 170 °C under 50 kN load. The prepared sheets were crosslinked in hot water at 90 ± 1 °C for 20 h. After crosslinking, the sheets were dried in vacuum oven for 16 h at 70 °C.
Particle Size Distribution
Particles size distribution of CMC was calculated by Malvern Instrument (SB.0D) using ethanol as dispersant medium rather than water because CMC is soluble in water.
Structural Analysis
The structure analysis of the blends was examined by Fourier transform infrared spectroscopy (FTIR). The IR spectra of the films were recorded by FTIR spectrophotometer (Thermo Electron Corporation, Nicolet 6700) using attenuated total reflection technique. The spectrum was scanned from 4000 to 500 cm−1 at the resolution of 6 cm−1. An average of 32 scans was recorded.
Thermogravimetric Analysis
The thermal stability of the samples was studied by using thermogravimetric analysis. These experiments were performed on a Mettler Toledo (TGA/DSC star system), under nitrogen flow of 50 ml/min. Approximately 8–10 mg of sample was placed in the alumina pan at a heating rate of 10 °C/min from room temperature to 550 °C.
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) of the blended material was performed using DSC Q2000 instrument. The samples were cut into small pieces and ~5 mg of each sample was used for analysis. To remove the thermal history, the sample was heated from 25 to 180 °C and cooled to −50 °C at a cooling rate of 10 °C/min and then reheated up to 180 °C at a heating rate of 10 °C/min under nitrogen atmosphere.
Rheological Analysis
The time and temperature dependent storage modulus G′, loss modulus G″ and complex viscosity (η*) were determined by Advance Rheometric Expansion System (ARES) using parallel plate geometry having plate diameter of 25 mm. The specimens from the compression molded sheets were cut according to the diameter of the plate. The experiment was performed at 150 °C over the frequency range of 0.05–100 rad/s. The gap between the plates was automatically adjusted by the instrument.
Tensile Properties
The tensile properties were determined using an Instron tensile tester (model 5543). The instrument was operated at a crosshead speed of 50 mm min−1 using 5-kN static load cell. The specimens were cut into dumb bell shape (Dimension; Type 4, Standard; ISO 37:1994) from a 1 mm thick compression molded sheet. Five specimens from each sample were tested.
Creep Test
The creep experiments were performed using DMTA IV (Rheometric Scientific). The instrument was operated in tensile mode at 60 °C. The dimensions of the specimens were 1.4 mm × 6 mm × 25 mm. The constant static stress of 2 MPa was applied for 5 h. Five specimens from each sample were tested.
Morphological Analysis
Scanning electron microscope (Jeol, JSM, 6400F) was used to investigate the morphology of the prepared samples. Specimens were dried in vacuum oven for 24 h and then freeze fractured using liquid nitrogen. Carbon fibers were used to coat the fractured surfaces. The instrument was operated at 10–15 kV. Energy dispersive spectroscopy (EDS) was used to determine the elemental composition of the specimens.
Result and Discussion
Mechanism of Silane Crosslinking
The mechanisms of silane grafting and crosslinking reactions during and after processing are shown in Schemes 1 and 2. At 150–170 °C temperature, the free radical initiator, dicumyl peroxide dissociates into peroxy radicals [Scheme 1a]. These peroxy radicals then attach on C–H bond of polyethylene chain and abstract hydrogen to give polymer free radical [Scheme 1b]. The combination of free radicals give radical induced crosslinking of polyethylene chain [Scheme 1c]. The double bond of VTES which is also susceptible for the radical attack was responsible for the grafting of VTES on polyethylene backbone as shown in Scheme 1d.

Reactions during processing: generation of free radicals from dicumyl peroxide (a), abstraction of hydrogen from polyethylene chain (b), crosslinking induced by free radicals (c), silane grafting reaction (d)

Reactions during crosslinking: hydrolysis (a), self condensation of silanol moieties (b), condensation of silanol with OR group of carboxymethyl cellulose (c)
The silane crosslinking reactions of polyethylene were carried out in boiling water. The first step during crosslinking was the hydrolysis of the silane into the silanol as shown in Scheme 2a. It has been reported in the literature that silane crosslinking reaction initiates during processing [23] as CMC already contains some moisture, which causes hydrolysis of alkoxy groups. The second step was the condensation of the silanol groups of the grafted polyethylene which is termed as self condensation reaction [see Scheme 2b]. The silanol groups may also condense with alkoxy group of CMC, thus, forming an ether linkage which connects the polyethylene with CMC via siloxane bond [Scheme 2c]. The presence of above-mentioned linkages was confirmed by FTIR analysis [Sect. 3.3].
Particle Size Distribution
The particle size distribution of CMC is shown in Fig. 1. The particle size varies approximately from 0.8 to 90 μm and the average particle size is in the range of 8 μm to 40 μm. The particles with sizes between 0.8 and 3 μm consist of less than 10% of the total volume. The particles with size between 3 and 8 μm contribute 30% by volume, whereas the particles with sizes between 8 and 40 μm contribute 40% by volume.

Particle size distribution of carboxymethyl cellulose
Structural Analysis
Figure 2 shows FTIR spectra of virgin PE, non-crosslinked (HC30) and crosslinked (XHC30) blends. The important absorption peaks of siloxane appeared in the range of 800–1200 cm−1. The XHC30 showed characteristic absorption peaks of (Si–O–Si) at 1050 and 1021 cm−1 [24, 25]. The symmetric vibration of (Si–O–Si) was also observed at 812 cm−1 which was absent in the non-crosslinked HC30 and virgin PE samples. The characteristic absorption peaks of CMC were found in both non-crosslinked and crosslinked formulations. The stretching frequencies of carboxyl and hydroxyl group appeared at 1581 and 3390 cm−1, respectively. The important absorption band appearing at 1159 cm−1 was assigned to Si–O–C, which reflects the crosslinking reaction [26].

FTIR spectra of HDPE/CMC blends
Thermogravimetric Analysis
Figure 3 shows weight loss of HDPE/CMC blends against temperature and Table 2 summarize thermal degradation temperature for major weight losses. The virgin PE and XPE samples showed single stage of degradation in the range of 410–500 °C. The silane crosslinked XPE exhibited higher thermal stability than virgin PE. The thermograms for non-crosslinked HDPE/CMC blends showed three stages of degradation. The first stage ranging from 90 to 150 °C was attributed to the loss of water present in CMC due to its hygroscopic nature. The second stage ranging from 250 to 410 °C was due to thermal degradation of CMC. During this stage, dehydration, ring scission and decomposition of the CMC occurred [27]. The third stage (ranging from 410 to 500 °C) was attributed to the decomposition of HDPE as observed in case of virgin PE. The crosslinked HDPE/CMC blends also showed three stages of degradation. The first stage ranging from 90 to 150 °C was attributed to the loss of water for both non-crosslinked and crosslinked blends. The second and third stages of degradation were similar for the non-crosslinked and crosslinked blends.

TGA thermograms of HDPE/CMC blends
Table 2 shows weight loss and weight residues of non-crosslinked and crosslinked blends at 150, 410 and 500 °C. At 150 °C, the moisture contents of both types of blends were completely lost but comparatively the non-crosslinked blends shows greater weight loss. The degradation of virgin PE and crosslinked XPE was not observed at 150 °C. The decomposition of CMC started at 410 °C. The major weight loss was observed for the non-crosslinked blends HC30 and HC50, which are 18.4 and 13.4%, respectively. At 410 °C, the crosslinked blends XHC30 and XHC50 showed lower weight loss of 10 and 4.7%, respectively. The minimum weight residue (3%) was noticed for virgin PE and the maximum weight residue (16%) was observed for XHC50 at 500 °C. This higher percentage of weight residue of crosslinked blends are mainly because of the compatibility between HDPE and CMC due to both free radical and condensation reactions.
Differential Scanning Calorimetry
Table 3 shows melting temperature (Tm), heat of fusion (∆Hf) and the percentage crystallinity of HDPE/CMC blends. Almost no difference was observed among the Tm values of non-crosslinked and crosslinked formulations but the percentage crystallinity differed to an appreciable extent. Comparatively, the crosslinked samples showed lower crystallinity than the non-crosslinked samples. The virgin PE has 63.4% crystallinity, whereas XHP has 61.7% and the observed difference was 1.7%. Similarly, the percentage crystallinity of HC40 and HC50 were 41.8 and 41.1% while that of XHC40 and XHC50 were 40.4 and 39.2%, respectively. In this case, there is a total reduction of 1.4 and 1.9%, respectively. As compared to PE, the addition of 20 phr of CMC reduced its crystallinity from 63 to 52%, while at 40 and 50 phr CMC loading, same percentage of crystallinity was observed. The lowering of percentage crystallinity for crosslinked formulations was due to the formation of network structure [28]. The network reduced the chain flexibility which reduced percentage crystallinity. Similar tendency has been previously observed in thermoplastic wood blends [24].
Rheological Analysis
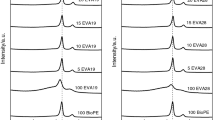
Figures 4 and 5 give the dynamic shear moduli of non-crosslinked and crosslinked formulations at 150 °C and the complex viscosities of these formulations are shown in Fig. 6. In the case of non-crosslinked formulations (Fig. 4), the G″ curves were observed at higher values than the G′ curves. This behavior was exhibited by blends having poor interaction between the components interphase as the components are weakly associated with each other. On the other hand, an opposite behavior was observed for the crosslinked formulations and now the G′ values were higher than G″ which means a strong interaction between the blended polymers interface. The components are strongly associated to each other and reflect the elastic nature of the material. In fact, the silane crosslinker connected the blended polymers which enhanced the elastic properties of these materials.

Dynamic shear moduli (G′ and G″) of non-crosslinked HDPE/CMC blends at 150 °C

Dynamic shear moduli (G′ and G″) of crosslinked HDPE/CMC blends at 150 °C

Complex viscosities (η*) of HDPE/CMC blends at 150 °C
The second important rheological feature of both crosslinked and non-crosslinked formulations was the increase in G′ and G″ values with increase in frequency. This was due to the fact that at low frequency, time is large enough and the entangled chains relax slowly. This slow relaxation tends to reduce the G′ and G″ values. However, when the polymer chains were deformed at higher frequency, the entangled chains have less time to relax back which increases the moduli. This behavior was first reported by Baghaei et al. [29] for the compatibilized LDPE/POE blend.
The Figs. 4, 5 and 6 revealed that both the dynamic shear moduli and the complex viscosities of the crosslinked and the non-crosslinked formulations increased with increasing amount of filler loading. The G″ values of non-crosslinked formulations increased step-wise with increase in CMC loading. The increase in G″ values with filler loading corresponded to weak interaction between the blended polymers interface. As CMC is hydrophilic and polyethylene is hydrophobic in nature, therefore, the resultant blends of these two polymers were immiscible. This was the main factor which enhanced the G″ values at different filler loadings in non-crosslinked formulations. In Fig. 5, the silane crosslinked formulations show opposite behavior. Instead of G″, G′ values gradually increased with higher loadings. This behavior corresponds to strong interaction between blended polymers interface due to silane crosslinking.
Figure 6 shows complex viscosities of non-crosslinked and crosslinked HDPE/CMC blends. The crosslinked blends showed higher complex viscosities which were attributed to the formation of network structure due to silane crosslinking. The network structure causes greater resistance to the applied stress.
The complex viscosity of both non-crosslinked and crosslinked blends also depended upon the amount of filler. At higher filler loading, higher η*values were observed for both types of blends. Figure 6 shows that η* values were also frequency dependent and decreased with increasing operating frequency. This behavior is called shear thinning effect in molten state [28].
Tensile Properties
Table 4 shows the tensile strength (TS) and elongation at break (Eb) values of non-crosslinked and crosslinked blends. It was observed that TS of crosslinked samples was much higher than the non-crosslinked samples. The Eb values showed a decreasing trend for both types of blends. The tensile strength of virgin PE and XPE were 8.1 MPa and 9.3 MPa, respectively. The tensile strength of XHP was increased up to 12.9%. Similarly, the XHC20 (16.1 MPa) showed 60.2% enhancement as compared to HC20 (6.4 MPa). The maximum tensile strength was observed for XHC40 (19.6 MPa) which was 61.7% greater than HC40 (7.5 MPa). In contrast, TS of non-crosslinked and crosslinked blends showed a decreasing trend at 50 phr CMC loading because at higher filler loadings the interactive forces between hydrophobic and hydrophilic chains become less effective. The higher TS of XHC50 as compared to HC50 was attributed to the compatibility between the HDPE and CMC due to both free radical and condensation reactions.
Both the non-crosslinked and crosslinked blends showed decreasing trend of Eb values but this effect is well-defined in the non-crosslinked formulations. The maximum Eb value was observed for XHC20 which is 19.1%, while HC20 showed 13.0%. The percentage elongation values decreased with higher CMC loading. Thus, HC50 and XHC50 show minimum Eb values which are 9.6 and 11.7%, respectively. This behavior is also reported in the literature [30, 31].
Creep Test
Figure 7 shows-short term creep test for both non-crosslinked and crosslinked formulations. Short-term creep experiments were carried out by dynamic thermal mechanical analyzer (DTMA). The creep analysis was carried at 60 °C for 5 h under the static mode.

Creep analysis of non-crosslinked and crosslinked HDPE/CMC blends at 60 °C
The non-crosslinked blends showed greater deformation as compared to crosslinked blends. The virgin PE had highest deformation as compared to XPE. It can be seen that deformation of these blends decreased with increasing amount of CMC loading. The least deformation was observed for XHC50 and HC50. The crosslinked blends showed minimum deformations, which is attributed to both free radical and condensation reactions. The crosslinking holds the macromolecular chains firmly whereas, in non-crosslinked blends, the molecular chains are free to move and the hydrophobic-hydrophilic interactions between HDPE and CMC further facilitate this phenomenon.
Morphological Analysis
Figure 8a, b shows SEM images of the fractured surfaces of non-crosslinked (HC30) and crosslinked (XHC30) blends. The particle size of CMC in non-crosslinked sample (HC30) is greater than 10 µm and is weakly bound to the polyethylene. Wide gaps are visible between PE and CMC interface, which indicates weak interaction that caused lowering of mechanical properties. The crosslinked formulation (XHC30) showed strong adhesion between the polymers interface. This behavior was attributed to both condensation and free radical reactions that firmly bound the two immiscible components.

Scanning electron micrographs of HDPE/CMC blends: HC30 (a, b), XHC30 (c, d)
Conclusions
The physical properties of HDPE/CMC blends can be greatly enhanced both by condensation and free radicals reactions. These reactions were confirmed by FTIR analysis. These reactions were attributed to the thermal stability and compatibility of the blends. The maximum tensile strength of 19.6 MPa was observed for the crosslinked blend having 40 phr CMC loading. Similarly, the compatibility and strong adhesion of blended polymers were also observed in short term creep deformation.
References
Orhan Y, Hrenovic J, Buyukgungor H (2004) Biodegradation of plastic compost bags under controlled soil conditions. Acta Chim Slov 51:579–588
Tzankova DN, Lamantia FP (1999) Recycling of the light fraction from municipal post-consumer plastics effect of adding wood fibers. Polym Adv Technol 10:607–614
Constance R, Chu AZ, Monosov DA (1995) In situ assessment of cell viability within biodegradable polylactic acid polymer matrices. Biomaterials 16:1381–1384
Maria R, Mariola J, Helena J (1998) Biodegradation of polycaprolactone in seawater. React Funct Polym 38:27–30
Alain B, Sylvie P (2008) Investigations on mechanical properties of poly (propylene) and poly (lactic acid) reinforced by miscanthus fibers. Appl Sci Manuf 4:1444–1454
Otey FH, Westhoff RP, Doane WM (1988) Biodegradable starch-based blown films, industrial and engineering chemistry products. Res Dev 19:592–595
Magnus B, Paul G, Kristiina O (2005) The effect of crosslinking on the properties of polyethylene/wood flour composites. Compos Sci Technol 65:1468–1479
Araujo JR, Waldman WR, De Paoli MA (2005) Thermal properties of high density polyethylene composites with natural fibres coupling agent effect. Polym Degrad Stab 93:1770–1775
Girones J, Pimenta MTB, Vilaseca F, de Carvalho AJF, Mutje P, Curvelo AS et al (2007) Blocked isocyanates as coupling agents for cellulose-based composites. Carbohydr Polym 68:537–543
Hongdian L, Yuan H, Ming L, Zuyao C, Weicheng F (2006) Structure characteristics and thermal properties of silane-grafted-polyethylene/clay nanocomposite prepared by reactive extrusion. Compos Sci Technol 66:3035–3039
Valenza A, Visco AM, Acierno D (2002) Characterization of blends with polyamide 6 and ethylene acrylic acid copolymers at different acrylic acid content. Polym Test 21:101–109
Racz I, Dea KA, Borsa J (1995) Fibrous carboxymethylcellulose by pad roll technology. Text Res J 1995(65):348–354
Mancinia MF, Nobilia F, Tossicia R, Wohlfahrt-Mehrensb M, Marassia R (2011) High performance, environmentally friendly and low cost anodes for lithium-ionbattery based on TiO2 anatase and water soluble binder carboxymethyl cellulose. J Power Sources 196:9665–9671
Zhang PN, Ross R, Kostecki F, Kong S, Sloop JB, Kerr K, Striebel EJ, Cairns F, McLarnon J et al (2001) Electrochem. Soc 148:A463
Damen J, Hassoun M, Mastragostino B, Scrosati J (2010) Power Sources 195:6902
Kim GT, Jeong SS, Joost M, Rocca E, Winter M, Passerini A et al (2011) Balducci. J Power Sources 196:2187
Fransson L, Eriksson T, Edström K, Gustafsson T, Thomas J (2001) Power Sources 101:1–9
Yan Baoa B, Jianzhong M, Li N (2011) Synthesis and swelling behaviors of sodium carboxymethyl cellulose-g-poly(AA-co-AM-co-AMPS)/MMT superabsorbent hydrogel. Carbohydr Polym 84:76–82
http://drugtopics.modernmedicine.com/drugtopics/Top+News/Overview-of-pharmaceutical-excipients-used-in-tabl/ArticleStandard/Article/detail/561047. Accessed 19 Aug 2011
Sayed S, Mahmoud KH, Fatah AA, Hassen A (2011) DSC, TGA and dielectric properties of carboxymethyl cellulose/polyvinyl alcohol blends. Phys B 406:406–4076
Xiaofei M, Peter R, Chang B, Jiugao YA (2008) Properties of biodegradable thermoplastic pea starch/carboxymethyl cellulose and pea starch/microcrystalline cellulose composites. Carbohydr Polym 72:369–375
Sadullah M, Tariq Y, Peter H, Humaira MS, Timothy N (2011) Thermal, rheological, mechanical and morphological behavior of HDPE/chitosan blend. Carbohydr Polym 83:414–421
Arkles B, Steinmetz JR, Zazyczny J, Mehta P (1992) J Adhes Sci Technol 6:193–206
Magnus B, Oksman K (2006) The use of silane technology in crosslinking polyethylene/wood flour composites. Appl Sci Manuf 7:52–765
Zhengzhou W, Yuan H, Zhou G, Rouwemn Z (2003) Halogen-free flame retardation and silane crosslinking of polyethylenes. Polym Test 22:533–538
Zakirov AS, Khabibullaev PK, Choi CK (2009) Structural characterization and electro-physical properties for SiOC(-H) low-k dielectric films. J Phys B 8:260–278
Nadia R, Tariq Y, Zareen A (2008) Synthesis of carboxymethyl-CMC/acrylic acid hydrogel using silane crosslinker. E-Polymers 8:142–157
Jiao C, Wang Z, Liang X, Hu Y (2005) Non-isothermal crystallization kinetics of silane crosslinked polyethylene. Polym Test 24:71–80
Baghaei B, Jafari SH, Khonakdar HA, Rezaeian I, Ashabi L, Ahmadian S et al (2009) Interfacially compatibilized LDPE/POE blends reinforced with nanoclay: investigation of morphology, rheology and dynamic mechanical properties. Polym Bull 62:255–270
Byung CJ, Soo YH (2011) Mechanical properties and morphology of the modified HDPE/starch reactive blend. J Appl Polym Sci 82:3313–3320
Balsuriya PW, Mai YW, Wu J (2005) Mechanical properties of wood flake polyethylene composite. II. Interface modification. J Appl Polym Sci 83:2505–2521
Acknowledgements
The author wishes to thank Higher Education Commission of Pakistan for financial assistance and the Center for High Performance Polymer, University of Queensland, Australia for technical support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mir, S., Yasin, T., Siddiqi, H.M. et al. Thermal, Rheological, Mechanical and Morphological Behavior of High Density Polyethylene and Carboxymethyl Cellulose Blend. J Polym Environ 25, 1011–1020 (2017). https://doi.org/10.1007/s10924-016-0880-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-016-0880-6