
Abstract
In this work, keratin was extracted from chicken feather waste via an environment-friendly method, sulphitolysis method, by using various sodium metabisulphite contents (0.0–0.5 M). Percentage yield and molecular weight of the extracted keratin were characterized by gravimetry and gel electrophoresis (SDS-PAGE), respectively. It was found that the yield increased, with the increase of sodium meta-bisulphite content, to the maximum value of 87.6 % yield, by using sodium meta-bisulphite content at 0.2 M. Molecular weight range of the product also decreased with the increase of the sodium meta-bisulphite content. The extracted keratin, with highest molecular weight ranged between 12 and 20 kDa, was further used for fabricating into fibers by using the electrospinning process. It was found that pure keratin solution could not be electrospun into fiber. However, by blending keratin with more than 10 wt% PLA, the fibers can be prepared. Results from FTIR and DSC also reveal that the crystal structure of the keratin changed from the β-sheet structure (rigid and small displacement characteristic) to the α-helix structure (elastic and large displacement characteristic) after keratin/PLA blends (10–50 wt% keratin) were prepared by the electrospinning method.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Chicken feather waste from the poultry industry, which is accounted for about 3600 million tons per year world-wide, is considered to be a critical problem in many countries, including Thailand [1]. In general, the chicken feather waste can be eliminated by several methods such as disposals, burning and burying. These methods, however, are environment-unfriendly. Therefore, the modification of the chicken feather waste for use as value added products in many applications such as tissue engineering, composites and filters are interesting [2–4]. The chicken feather waste consists of about keratin proteins (91 %), fat (1.3 %) and water (7.7 %) [5]. About 18–20 types of amino acids can be found in keratin. These amino acids are bonded together via peptide bonds. Each amino acid contains different reactive sites such as hydroxyl group, carboxylic groups, amine group, thiol group and aromatic group. In addition, the amino acids in keratin chains are both intramolecular and intermolecular interacted by disulphide bonds, ionic bonds, hydrogen bonds or hydrophobic bonds. From these reasons, the extraction of keratin requires a range of chemical substances to break these bonds [6].
In term of applications, keratin can be used as raw material for preparing scaffolds for tissue engineering and heavy metal ion filters due to its biocompatibility, good cell adhesion and high polarity. For these products, the polymeric materials are usually utilized in a form of nanofibers, and the fibers can be prepared by several techniques including, drawing [7], template synthesis [8], phase separation [9], self assembly [10] and electrospinning [11]. In this study, the preparation of nanofiber via electrospinning is of interest, owing to the fact that the fabrication process can be scale up [11]. However, the electrospinning of keratin is not straightforward, due to the fact that molecular weight of keratin is not sufficiently high [12, 13]. Several attempts have been made to increase the molecular weight of protein by connecting molecular chains together using some crosslinking agents such as hexamethydiisocyanate [14], glutaraldehyde [15], citric acid [16, 17] and glyoxal [18]. These chemicals are harmful for tissue engineering applications, complex processes, therefore, are needed to produce the crosslinked proteins. Alternatively, specific polymers such as poly(ethylene oxide) (PEO) and nylon, have been greatly selected for blending with keratin to improve the electrospinning processability of keratin [13, 19–24]. However, these polymers are synthetic, water soluble and non-biodegradable, which are not suitable for filter and scaffold applications. In this study, poly(lactic acid) (PLA) was selected to blend with keratin. This is because PLA is biocompatible and biodegradable, making suitable for the scaffold application. In fact, the preparation of PLA nanofibers via electrospinning for the filter application has been reported [25].
In terms of extraction, effects of chemicals concentration for extracting keratin from wool using a sulphitolysis method have been previously reported [19]. The optimum chemical concentration for extraction has also been reported. However, the content of amino acids and cystine in wool and chicken feather are different. Chicken feathers contain lower amount of cystine (8.2 %), as compared to that in wool (14.4 %) [26]. This suggests that lower disulphide bond in keratin extracted from chicken feathers should be used and the optimum condition can be different.
In this study, the extraction of keratin and feasibility of preparing keratin-based nanofibers via electrospinning are of interest. The aims of this work were to investigate the optimum concentration of sodium-metabisulphite, for extracting keratin from chicken feathers using the sulphitolysis method. Afterwards, the properties of the keratin, PLA and keratin/PLA blend solutions, such as viscosity and conductivity, were investigated. Finally, the effect of processing conditions and keratin blend ratio on the secondary structure of keratin was also interested.
Experimental
Chemicals
Chicken feathers were received from Better Foods Company Limited (Thailand). Ethanol (ACS reagent grade) used as disinfectant was supplied from Merck (Germany). Petroleum ether (ACS reagent grade) was supplied from J.T.Baker® (USA). Sodium dodecyl sulphate (analysis grade) and sodium hydroxide (ACS reagent grade) were purchased from Carlo Erba Reagents (France). Sodium metabisulphite (analytical reagent grade) and urea (analytical reagent grade) were supplied from Ajax Finechem Pty Ltd (Australia). Nylon filters (50 and 150 μm) were purchased from Plastech marketing Company Limited (Thailand). Formic acid (98–100 % ACS reagent grade) was supplied from Merck (Germany). Chloroform (analytical reagent grade) was supplied from RCI Labscan Limited (Thailand), and acetone (ACS reagent grade) was supplied from Merck (Germany).
Chicken Feather Preparation
Chicken feathers were cleaned using the following steps. The chicken feathers were firstly immersed in ethanol for 24 h, and then thoroughly washed with detergents and water respectively. The cleaned wet chicken feathers were then dried in an oven at 60 °C for 45 h. Next, the quills of the cleaned chicken feather were cut off, and the remaining parts of the feathers (the vanes and barbs) were then grinded by using a Cyclotech 1093 Sample mill. Short feathers with a length of about 0.6 mm can be obtained. The feathers were chemically treated in a soxhlet device with petroleum ether for 12 h to remove fatty matters, followed by washing with distilled water and drying at room temperature. The cleaned short feathers were then stored in closed containers.
Keratin Extraction
The cleaned short feathers (7 g) were immersed in 250 ml of aqueous solutions shown in Table 1, and continuously stirred at 65 °C for 5 h. Then, the extracted solutions were passed through a nylon mesh (150 and 50 μm respectively). The filtrates were subsequently dialyzed in a dialysis tube (MWCO 6000–8000 Da) with distilled water for 3 days (distilled water was changed 3 times a day). The solutions were subsequently concentrated using a rotary evaporator (Heidolph, Laborota 4003 Control) at 40 °C and 45 mBar. The prepared keratin was freeze-dried until its weight remained constant.
In term of extraction via sulphitolysis step, ionic bonds, hydrogen bonds, hydrophobic bonds and disulphide bonds were cleaved with sodium hydroxide, urea, sodium dodecyl sulphate and sodium metabisulphite respectively. In this work, sodium metabisulphite with a variety of concentration was used to break covalent disulphide bonds (Table 1). In this extraction step, the cysteine disulphide bonds in the keratin structure are cleaved with sulphite, and then cysteine thiol and cysteine-S-sulphonate (or “Bunte slat”) are released [19] as shown in reaction (1) Besides, the keratin yields could be calculated using Eq. (1).

Preparation of Keratin and Keratin/PLA Nanofibers
1 g of keratin was dissolved in formic acid (98 %) to prepare 10 wt% keratin solution, and 1 gof PLA was dissolved in chloroform and acetone (70/30 v/v) to prepare the 10 wt% PLA solution. The keratin/PLA solution was prepared by mixing the keratin solution with the PLA solution under mechanical stirring at room temperature for 24 h. Keratin nanofibers and keratin/PLA nanofibers were produced using the electrospinning method with the operating condition, as shown in Table 2.
Characterizations
The separation of protein molecular weights was carried out by using a sodium dodecyl sulfate-polyacrylamide gel electrophoresis technique (SDS-PAGE). The sample was dissolved in a buffer containing SDS, tris–HCl buffer (pH 6.8), dithiothreitol (DTT), glycerol and bromophenol blue. The solution was heated in a boiling water bath for 5 min. The stacking and the separating gel were 12 % polyacrylamide. Electrophoresis voltage used was 200 V. After electrophoresis, the gel was washed with water and then stained with a staining solution (Coomassie Brilliant Blue R-250, BIO-RAD). Subsequently, the sample was de-stained with a mixed solution (water, methanol, glacial acetic acid). The protein standard (Precision plus protein all blue standards, BIO-RAD) was used for calibration.
The viscosity of the solutions was measured using a RotoVisco1, cone and plate rheometer, a cone-plate geometry which has a diameter of 60 mm, an angle of 1° and a gap of 0.047 mm, equipped with a temperature control device at 25 ± 1 °C. The shear rate increased from 50 to 1000 s−1. In addition, the ion conductivity of the solutions was measured using a Proline conductivity meter B250.
The morphology of nanofibers was investigated using a Joel scanning electron microscope (JSM-6610LV) equipped with a secondary electron detector under an accelerating voltage of 10 kV. The average fiber diameters of the nanofibers, and standard deviation values were then investigated from at least 100 nanofibers.
Chemical structures of neat keratin and keratin/PLA nanofibers were characterized by using an ATR mode Fourier transform infrared spectroscopy (Thermo Scientific™ Nicolet™ iS™5 FT-IR Spectrometer). The samples were scanned over the wavenumber ranging between 550 and 4000 cm−1.
Thermal behaviors of keratin and keratin/PLA nanofibers were studied using a differential scanning calorimetry (DSC) coupled with Netzsch DSC 204 thermal analyze. About 5 mg of the sample was placed in an aluminium pan. DSC scanning was performed over the temperature ranging between 30 and 350 °C. The heating rate of 10 °C min−1 and the nitrogen flow rate gas of 20 ml min−1 were used.
Results and Discussion
Extracted Keratin Characterization
Yields of Extracted Keratin from Chicken Feathers (CF)
The yield of keratin obtained from chicken feathers treated with different concentrations of sodium metabisulphite was shown in Fig. 1. The highest yield (~87.6 %) was obtained when 0.2 M of sodium metabisulphite was used. A lower yield (~36.2–62.9 %), on the other hand, can be found at a lower sodium metabisulphite concentration (0.0 and 0.1 M). This is because a low concentration of sodium metabisulphite cannot break the agglomerated macro molecules of keratin. The agglomerated molecules, therefore, were unable to pass through the filter, whereas only unfolded keratin chains were capable of passing through the filter. This resulted in the low yield of keratin. Moreover, when the concentration of sodium metabisulphite was increased above 0.2 M, the keratin yield decreased. This is due to the fact that most of the disulphide bonds are broken, resulting in short keratin chains. These short keratin chains can be transferred from a dialysis tube to outside.

Keratin yields from the sulphitolysis extraction with various sodium meta-bisulphite concentrations
Molecular Weights of Extracted CF-Keratins
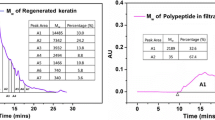
SDS-PAGE patterns and the average molecular weight values of the keratin extracted under different sodium metabisulphite concentrations are shown in Fig. 2, and Table 3, respectively. It was found that molecular weights of the products decreased with the increase of concentration of sodium metabisulphite used. For example, the molecular weight ranges of 11–20 kDa were obtained when keratin was extracted with 0.2 and 0.3 M of sodium metabisulphite whereas the lower ranges (10–15 and 12–13 kDa) were obtained when 0.4 and 0.5 M of sodium metabisulphite were used. The optimum concentration of sodium metabisulphite for the extraction was found to be 0.2–0.3 M. The keratin yield which was extracted by used concentration of 0.2 M of sodium metabisulphite, was higher than used 0.3 M. In this regard, molecular weight of the keratin seemed to be shorter due to the greater dissociation of covalent and disulphide bonds. Notably, the highest molecular weights of keratin prepared in this work was found to be two times higher than that of keratin prepared by O’Donnell (10 kDa) [27, 28]. Therefore, in this study, 0.2 M of sodium metabisulphite was selected for preparation of keratin nanofibers via electrospinning process.

SDS-PAGE of protein standard (1) and the various extracted keratin (2–5) using different amount of sodium metabisulphite (0.5, 0.4, 0.3 and 0.2 M, respectively)
Properties of Keratin, PLA and Keratin/PLA Solutions
Figure 3 shows viscosity values of various keratin and keratin/PLA solutions. The non-Newtonian behavior can be found from all solutions. The PLA solution had the highest viscosity, and showed an initial shear-thickening at the low shear rate region (between 1 and 40 s−1). This was because of a formation of “physical gel” which was subsequently transformed into “an interface film” at the surface of the PLA solution. After dropping the PLA solution onto a plate of cone-plate rheometer for a few minutes, the solvents (chloroform and acetone) on the surface of the droplet was evaporated, resulting in a formation of the interfacial film. At higher shear rate (between 40 and 1000 s−1), this mechanism was interrupted. Then, a Newtonian behavior was observed with high viscosity value (at about 0.19 Pa s) [29, 30].

Viscosity values of keratin, PLA and various keratin/PLA solutions (90/10, 70/30, 50/50, 30/70 and 10/90 w/w respectively)
Notably, the viscosity of the keratin/PLA solution (10/90 w/w) was high at a low shear rate (1 s−1), and showed a shear-thinning behavior over the shear rate ranging between 2 and 100 s−1. This was due to a hydrophobic effect occurring between keratin and PLA. This effect has been previously reported [26]. Furthermore, the viscosity value remained constant at about 0.15 Pa s as the shear rate further increased up to 1000 s−1. The solution still exhibited the Newtonian behavior. This was due to the fact that the higher the shear rate, the greater the solution homogeneity.
The keratin/PLA solution (30/70 w/w) showed the Newtonian behavior for the total range of the shear rate at viscosity of 0.12 Pa s. This viscosity was lower than that of both 10/90 w/w solution and pure PLA solution due to the smaller amount of PLA. However, the 70/30 w/w solution showed an initial shear-thickening at the low shear rate (between 1 and 30 s−1) owing to an evaporation of the solvent in the solution.
When the weight fraction of keratin in the solution was higher than 50 wt%, the viscosity remained constant, regardless of the shear rate. This behavior is similar to other proteins. This may be attributed to the low molecular weights of proteins as compared to other polymers [29, 31]. Figure 4 shows the conductivity of the keratin/PLA solutions as a function of the keratin content. Conductivity of the solutions increased with the keratin content. This can be attributed to the presence of polar amino acid existing in the keratin [32].

Conductivities of keratin/PLA solutions
Processability and Morphology of Keratin and Keratin/PLA Electrospuns
From our preliminary study, it was found that keratin solution (in formic acid) cannot be prepared into nanofibers via the electrospinning, regardless of the concentration used (7–50 wt%). This is due to the low molecular weight of keratin. Consequently, PLA with an average molecular weight of 200,000 g mol−1 was used as a kind of carrier polymer for blending with keratin prior to the electrospinning.
Figure 5 shows SEM images of various electrospun fibers and the average diameters of nanofibers were reported in Table 4. The morphology of the electrospun fibers was also affected by the amount of keratin in the solution. It is noted that only droplets with diameters of ~275 nm on the substrate can be observed when the large amount of keratin in the PLA solution (90 wt%) was prepared (Fig. 5a) because of low chain entanglement and very high conductivity of keratin. This caused very low surface tension of the solution at the end of tip. This made it unable to form a Taylor’s cone during electrospinning to prepare a fiber form. Only droplets were deposited on a ground collector. With the 30 wt% of keratin in the PLA solution, microfibers with droplets along the fiber surface could be found (Fig. 5b). This was due to the extensive content of the low viscosity keratin, and high conductive properties which can be phase separated easily during the electrospinning. The average diameters of fibers and droplets were about 921 and 721 nm, respectively. The continuous electrospun fibers with diameters of ~341 and 184 nm were found from the keratin/PLA solution of 50/50 and 30/70 w/w respectively (Fig. 5c, d). This was because of a sufficient molecular chain entanglements between keratin and PLA [20]. The keratin/PLA blended solution (10/90 w/w) was unable to be electrospun by using the needle with a diameter of 0.4 mm due to the blockage of PLA inside the needle tip causing the high viscosity of the solution. The needle with a diameter of 0.8 mm was subsequently used as a replacement. Notably, different fibers were fused together at the junctions due to the incomplete evaporation of the mixed solution in the short time (Fig. 5e) [11, 33]. The PLA porous fibers were shown in Fig. 5f. This is due to the fact that during the electrospinning process, chloroform with a higher evaporation rate was firstly evaporated, and acetone, which was trapped inside the fiber, was evaporated afterward [11].

SEM images of keratin/PLA electrospun fibers at various ratios a 90/10, b 70/30, c 50/50, d 30/70, e 10/90 and f neat-PLA, respectively
Analysis of Molecular Structure of Keratin/PLA Electrospun and Films
Figure 6 shows FTIR spectra of keratin films, keratin/PLA and neat PLA nanofibers. For the extracted keratin sample, the FTIR bands corresponding to amide A (amide stretching vibration, 3290 cm−1), amide B (amide stretching vibration, 3063–3200 cm−1), amide I (carbonyl stretching vibration, 1600–1700 cm−1), amide II (NH bending vibration, 1510–1580 cm−1), amide III (1220–1300 cm−1) and bunte salts (thiosulphate ion vibration, 1020 cm−1) can be observed. Similar spectrum was observed in related literature [20, 34, 35].

Overlaid FT-IR spectra of keratin film, keratin/PLA and neat PLA nanofiber mats
No keratin peaks could be observed from the keratin/PLA fibers (10/90 w/w), probably due to the lower amount of keratin. When the amount of keratin in the sample was increased to 50 wt%, however, the higher intensity of the adsorption bands could be clearly observed [20].
From the above results, the continuous fibers prepared from the keratin/PLA solution (50/50 v/v) were further studied, as it contained the highest amount of keratin. The EDS dot mapping technique was used to monitor distribution of amide group of keratin, using Kα X-ray signal of N atom (Fig. 7). It was found that the signal was fully distributed along the electrospun fiber mats.

Element analysis from EDS technique and the composition percentages of keratin/PLA (50/50) nanofiber mat a SEM image and b N elements
Analysis of Secondary Structure of Keratin and Keratin/PLA Samples
Normally, keratin comprised two types of crystal structures: α-helix and β-sheet. The α-helix structure is a right-hand helical polypeptide chain with 3.6 amino acids per turn of the helix. It has intramolecular hydrogen bonding, contributing to elastic and flexible properties of the material. On the other hand, the β-sheet structure is a crystal structure with a pleated sheet form. Intermolecular hydrogen bonding between backbone molecules and the adjacent chains (parallel either or anti-parallel direction) also exist, providing a more rigid structure than α-helix. This leads to high strength and small displacement characteristic of the material [36].
In this study, attempts were made to investigate the crystal structure of keratin in order to draw a relationship between structure and strength of keratin product. This can be monitored by using the FTIR technique. In general, the secondary structure of the proteins, corresponding to the amide I band located at between 1600 and 1700 cm−1 was observed from the FTIR spectrum. The bands located at ~1650 cm−1 and between 1610 and 1633 cm−1, corresponding to the α-helix structure and β-sheet structure respectively, can also be found in the FTIR spectrum of a neat keratin film [35, 37].
Figure 8a shows the FTIR spectrum of the keratin film and keratin/PLA nanofiber mats. The higher β-sheet band area (located at ~1623 cm−1) can be seen from the keratin film. This was because the molecular chains of keratin had enough time to rearrange and form the β-sheet structure. Keratin/PLA nanofiber mats (50/50 w/w) (Fig. 8b), on the other hand, showed the decrease of the β-sheet structure band area, and the increase of the α-helix band area. This suggests that the molecular chains of keratin may be hindered by PLA chains. Similar effect was observed by FTIR spectrum of keratin/poly(ethylene oxide) blend nanofiber in a study on structure and properties of keratin/Poly(ethylene oxide) blend nanofibers [20].

Curve-fitted amide I peaks of a keratin film and b keratin/PLA 50/50 nanofiber mat
Figure 9 shows DSC thermograms of keratin film, keratin/PLA and PLA fiber mat, and Table 5 summarizes the DSC results obtained from the first heating. Compared to PLA fiber mat, the cold crystallization of keratin/PLA blend fibers occurred at much lower temperature. ΔH cc of the blend fibers were lower than that of the neat PLA. In addition, the keratin/PLA fibers showed a higher ΔH m than the neat PLA fibers. This indicates that the crystallization and crystallinity of PLA can be induced by keratin chains [23]. Interestingly, the values of ΔH m obtained from all electrospun fibers were higher than that of ΔH cc. This indicates that PLA can be crystallized during the electrospinning process because the PLA solution was rapidly ejected from Taylor’s cone to the ground collector, aligning molecular chains of PLA.

DSC thermograms of a keratin film, nanofiber of keratin/PLA blended at the ratio of b 50/50, c 30/70, d 10/90 and e PLA nanofiber mats
All of the keratin/PLA electrospun fibers show the slightly decrease of the glass transition temperature, compared to neat PLA fibers. The cold crystallization and melting temperature of keratin/PLA fibers were lower than those of neat PLA fibers. These are mainly due to the residual moisture and solvent in the blend solutions. The absorbed (or the residual) moisture allows PLA molecules to move easily, resulting in the lower crystallization and melting temperatures [23, 38].
The keratin film showed the endothermic peak at 100–150 °C, which can be related to the amount of water absorption, as shown in Fig. 9a. This is related to the use of energy to break hydrogen bonding between water and hydrophilic keratin surface [12, 31, 39, 40].
Change in the secondary structure of keratin in both films and keratin/PLA nanofiber mats (50/50 w/w), was shown in Fig. 10 and Table 6. The keratin films showed the higher β-sheet peak area (at 251–300 °C), compared to the α-helix peak area (at 206–250 °C). On the other hand, compared to pure keratin films, the keratin/PLA nanofiber mats (50/50 w/w) showed a higher area of the α-helix peak at 196–260 °C and a lower area of the β-sheet peak at 260–295 °C. Noteworthy, the area was reported after the actual content of keratin in the blend was corrected. The keratin chains and PLA chains were hindered each other, resulting in the disruption of molecular orientation and formation of β-sheet structure. Besides, another factor contributing to the better stability of the β-sheet structure of keratin, is the electrospinning process. During the electrospinning, the solvent was evaporated rapidly, and molecular structure of keratin cannot be rearranged into β-sheet structure within time [20].

DSC thermograms of the keratin denature temperature of a keratin films and b keratin/PLA (50/50) nanofiber mats
Conclusions
Keratin has been successfully extracted from chicken feathers using the sulphitolysis method. The molecular weight of keratin decreased with increase of sodium metabisulphite content. All blended solutions exhibited the non-Newtonian behavior. The keratin/PLA solution with the ratio of 10/90 w/w showed a different viscosity behavior. The viscosity was high at a low shear rate (1 s−1), and then exhibited a shear-thinning behavior over the shear rate range between 2 and 100 s−1. Furthermore, the viscosity remained constant (at about 0.15 Pa s) as the shear rate was increased up to 1000 s−1, and the Newtonian behavior was observed. The extracted keratin cannot be prepared into nanofiber via electrospinning. By blending with PLA, however, the keratin- based material can be electrospun. Specifically, when the keratin content in the solution was lower than 50 wt%, it can be successfully spun into continuous nanofibers. The electrospinning process was also capable of inducing the formation of the α-helix secondary structure, and destabilizing the β-sheet secondary structure of proteins. Last but not least, the cold crystallization and melting peaks of PLA in the blended keratin/PLA fiber mat (50/50) were observed, and the glass transition temperatures of PLA tended to decrease when keratin was added.
References
Barbut S (2002) Poultry products processing: an industry guide. CRC Press LLC, Boca Raton
Matthews JA, Wnek GE, Simpson DG, Bowlin GL (2002) Biomacromolecules 3:232
Hajra MG, Mehta K, Chase GG (2003) Sep Purif Technol 30:79
Ki CS, Gang EH, Um IC, Park YH (2007) J Membr Sci 302:20
Chinta SK, Landage SM, Krati Y (2013) IJIRSET 2:1158
Bhushan B (2010) Biophysics of human hair structural, nanomechanical, and nanotribological studies. Springer, Heidelberg
Ondarcuhu T, Joachim C (1998) Europhys Lett 42:215
Feng JJ (2002) Phys Fluids 14:3912
Ma PX, Zhang R, Biomed J (1999) Mater Res 46:60
Hartgerink JD, Beniash E, Stupp SI (2001) Science 294:1684
Ramakrishna S, Fujihara K, Teo W-E, Lim T-C, Ma Z (2005) An introduction to electrospinning and nanofibers. World Scientific, Singapore
Zoccola M, Aluigi A, Vineis C, Tonin C, Ferrero F, Piacentino MG (2008) Biomacromolecules 10:2819
Yuan J, Xing Z-C, Park S-W, Geng J, Kang I-K (2009) Macromol Res 17:850
Yao C, Li X, Song T (2007) J Appl Polym Sci 103:380
Selling GW, Woods KK, Sessa D, Biswas A (2008) Macromol Chem Phys 209:1003
Weijie X, David K, Wen Y, Yiqi Y (2008) Polym Int 57:1110
Jianga Q, Reddya N, Yang Y (2010) Acta Biomater 6:4042
Suwantong O, Pavasant P, Supaphol P (2011) Chiang Mai J Sci 38:56
Tonin C, Aluigi A, Vineis C, Varesano A, Montarsolo A, Ferrero F (2007) J Therm Anal Calorim 89:601
Aluigi A, Vineis C, Varesano A, Mazzuchetti G, Ferrero F, Tonin C (2008) Eur Polym J 44:2465
Katoh K, Shibayama M, Tanabe T, Yamauchi K (2004) J Appl Polym Sci 91:756
Yin G-B, Zhang Y-Z, Wu J-L, Wang S-D, Shi D-B, Fu W-G (2009) J Biomed Mater Res A 93:158
Zhang J, Jiang L, Zhu L (2006) Biomacromolecules 7:1551
Varesano A, Aluigi A, Vineis C, Tonin C (2008) J Polym Sci Polym Phys 46:1193
Juan L, Fei SX, Feng G, Qi LL, Rui C, Ying CC, Zhong Z (2014) Sci China Tech Sci 57:239
Graham CE, Waitkoff HK, Hier SW (1949) J Biol Chem 177:529
O’Donnell IJ (1973) Aust J Biol Sci 26:401
Arai KM, Takahashi R, Yokote Y, Akahane K (1983) J Biochem E 132:501
Sharma V, Jaishankar A, Wang Y-C, McKinley GH (2011) Soft Mater 7:5150
Neto WAR, Pereira IHL, Ayres E, de Paula ACC, Averous L, Góes AM, Oréfice RL, Bretas RES (2012) Polym Degrad Stab 97:2037
Um IC, Kweon HY, Park YH, Hudson S (2001) Int J Biol Macromol 29:91
Aluigi A, Corbellini A, Rombaldoni F, Zoccola M, Canetti M (2013) Int J Biol Macromol 57:30
Buchko CJ, Chen LC, Shen Y, Martin DC (1999) Polymer 40:7397
Yuan J, Shen J, Kang I-K (2008) Polym Int 57:1188
Surewicz WK, Mantsch HH, Chapman D (1993) Biochem US 32:389
Buehler MJ, Keten S (2008) Elast Nano Res 1:63
Byler DM, Susi H (1986) Biopolymers 25:469
Jiang L, Wolcott MP, Zhang J (2014) J Biomacromol 7:199
Paswan S (2012) Studying the arsenic absorption by keratin protein extracted from human hair. National Institute of Technology, Rourkela
Spei M, Holzem R (1987) Colloid Polym Sci 265:965
Acknowledgments
The authors are sincerely grateful to the Office of Higher Education Commission, strategic scholarships for a frontier research network program and the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission for providing a research grant to support this work (Project No. 56000558). We would also like to thank Ms. Thitiporn Teeravivattanakit, Asst. Prof. Dr. Chakrit Tachaapaikoon and Assoc. Prof. Dr. Khanok Ratanakhanokchai, Enzyme Technology Laboratory, Pilot Plant Development and Training Institute, KMUTT for supporting gel electrophoresis laboratory.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Isarankura Na Ayutthaya, S., Tanpichai, S. & Wootthikanokkhan, J. Keratin Extracted from Chicken Feather Waste: Extraction, Preparation, and Structural Characterization of the Keratin and Keratin/Biopolymer Films and Electrospuns. J Polym Environ 23, 506–516 (2015). https://doi.org/10.1007/s10924-015-0725-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-015-0725-8