
Abstract
Poly (vinyl alcohol) (PVA)/soy protein isolate (SPI) blend film plasticized with glycerol is fabricated by melting processing in presence of water. The structure and properties are investigated by Fourier-transform infrared spectroscopy, X-ray diffraction, differential scanning calorimeter, scanning electron microscope (SEM), mechanical testing, and thermo-gravimetric analysis (TGA). It is found that strong hydrogen bonds between soy protein and PVA macromolecules are formed. The incorporation of soy protein into PVA decreases the crystallinity of the latter. The crystallization and melting temperatures decrease with increasing SPI content. SEM shows that certain degree of phase separation occurs in the blend film, with soy protein phase finely dispersed in the PVA matrix. The PVA/SPI blend film possesses high flexibility even at high protein content, which ensures its potential applications as packaging film. TGA experiments shows that the incorporation of soy protein rendered the thermostability of the blend film.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biodegradable plastics can be easily degraded in the natural environment and is attracting more and more research interest [1–3], since the petroleum-derived plastics have caused worldwide environment problems. Agricultural resources, such as protein, starch, cellulose, are considered as substitutes to petroleum-based polymers because of its abundance, low cost, biodegradability, and good mechanical performance [4–6]. Soybean is one of the most abundant crops in the world. Soy protein, a by-product of soy oil industry, is regarded as a readily renewable biopolymer and a potential source for biodegradable materials [7, 8]. The commercial raw material of soy protein is soy protein isolate (SPI), which contains more than 90 % protein composed from 18 diverse amino acids [9]. In recent years, SPI has been used to fabricate food trays, fibers, containers, flatware, edible film, and packaging film [10, 11]. However, the neat SPI based products lack strength and rigidity, which inhibit their application in materials engineering [12, 13]. The common practices to resolve the problem mainly include adding plasticizers (polyols, hydroxylamine, amide, etc.) [14, 15], blending with other polymers [16], and/or reinforcing with fillers [17].
Poly (vinyl alcohol) (PVA) is a biodegradable polymer of petroleum source, which possesses excellent chemical resistance, optical/physical properties, and good film-forming capability [18–20]. Blending PVA with soy protein have been considered a reasonable choice to compensate the mechanical property weakness of soy protein products [21, 22]. In addition, soy protein is much cheaper than PVA, which would enlarge application of the blends. However, the PVA/SPI blend film is currently fabricated by solution casting, which is high energy consuming and inefficient.
In this work, the melt processing of PVA/SPI blend film was carried out, in which the plasticizers played an indispensable role. The structure and properties of the blend film were investigated. It was aimed to provide a facile and economic method to prepare biodegradable PVA/SPI blend film with excellent physical properties.
Experimental Section
Matetials
PVA (1799) was purchased from Kuraray Co., Ltd. (Japan). SPI (moisture ca. 6.5 wt%) was purchased from DuPont–Yunmeng Protein Technology Co. Ltd. (Yunmeng, China). Other chemicals were obtain from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China) and used as received.
Preparation of PVA/SPI Blend Film
Glycerol and water were mechanically stirred to prepare homogeneous solutions. SPI, PVA and the resulting solution were homogenized in a high-speed mixer (Beijing Huaxinke plastic machinery Co., Ltd, China) at room temperature. The glycerol content of all the samples was fixed at 40 phr (based on SPI + PVA). The resulted mixtures were charged into a HAAKE torque rheometer (TA Instruments, USA) and further mixed at 105 °C for 10 min with a stirring speed of 100 rpm. The mixture was subsequently compression molded on a plate vulcanizing machine (XLB-400, Huaqing Industry Group, Qingdao, China) to obtain a blend film. The molding temperature, pressure, and time were 110 °C, 5 ton, and 10 min, respectively. The obtained blend film was coded as PxSy, where x and y stands for the ratio of PVA and SPI. The content of SPI in the blends was kept lower than 50 %, due to its poor mechanical properties.
Characterization
Fourier transform infrared (FTIR) spectra of the samples were recorded on a Nicolet 5700 FTIR spectrometer (Thermo Electron Corporation, Waltham, MA, USA). The samples were prepared by mixing with fine powder of KBr and pressing. The spectra were obtained at a resolution of 4 cm−1 in the range from 4,000 to 400 cm−1.
X-ray diffraction (XRD) was recorded on a XRD instrument (XRD-6000, Shimadzu Co., Japan) using Cu–Kα radiation (0.154 nm) at 40 kV and 30 mA. XRD data were collected from 2θ = 5° to 40° at a scanning rate of 2 °/min.
Differential scanning calorimetry (DSC) was carried out on a DSC Q100 calorimeter (TA Instruments, USA) under nitrogen atmosphere. Samples of 5–10 mg in a pressure-sealed aluminum pan were maintained at 220 °C for 3 min to eliminate thermal history. Then the samples were quenched to 25 °C to obtain the crystallization curve, and then heated to 200 °C to obtain the melting curve with a heating rate of 20 °C min−1.
A scanning electron microscope (VEGA II, TESCAN, S.R.O, Czech Republic) was used to observe the cross-sections of fractured samples. Each sample was frozen using liquid nitrogen, fractured and then vacuum-dried to produce cross-sections. The fractured surfaces (cross-sections) of the plastics were sputtered with gold before observation.
Mechanical properties of the blend film were determined using a universal testing machine (Sans, Mts Systems China Co., Ltd) in tensile mode according to the ASTM D638 standard. Mechanical tensile data were averaged over at least five specimens. The samples were conditioned at 58 % RH before testing.
The kinetics of water absorption at room temperature (25 °C) was determined for all film. The specimens with dimensions around 10 × 10 × 0.2 mm3 were thin enough so that the diffusion was supposed to be unidirectional. The samples were first vacuum-dried overnight at 100 °C. After being weighted using a four-digit balance, the sample was conditioned in a desiccator containing NaBr saturated solution to ensure a RH of 58 %. The sample was removed at specific intervals and weighted. The water uptake (WU) was calculated by:
where m 0 and m t were the weights of the samples before and after a time t of conditioning, respectively. The mass of moisture absorbed at time t was expressed as [23]:
where m∞ was the weight of the sample at equilibrium, 2L was the thickness of the film, and D was the diffusion coefficient. At short times, this equation can be written as:
for (m t − m 0)/m ∞ ≤ 0.5, the relative difference between the diffusion coefficients calculated by Eqs. (3) and (2) was about 0.1 % [24]. The plots of [(m t − m 0)/m ∞]2 as a function of [4t/(πL 2)] were drawn for all samples with (m t − m 0)/m ∞ ≤ 0.5. The diffusion coefficients (D) were calculated from the slope of the plots.
Thermo-gravimetric analysis (TGA) was performed on a TGA Q5000 thermo-gravimetric analyzer from 25 to 600 °C at a heating rate of 10 °C/min under nitrogen atmosphere. The sample mass was about 5–10 mg.
Results and Discussion
FTIR Analysis
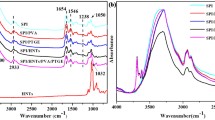
The FTIR spectra of PVA/SPI blend film were shown in Fig. 1. The wide absorption peaks at 3,300–3,400 cm−1 were attributed to the O–H bond stretching vibration. The peaks around 1,646, 1,541 and 1,240 cm−1 were attributed to the characteristic amide bands of soy protein: amide I (C=O stretching), amide II and III (N–H bending and C–N stretching), respectively.

FTIR spectra of PVA/SPI blend film
Neat PVA also exhibited C=O stretching peaks around 1651 cm−1, which was ascribed to the un-alcoholized acetate groups. The absorption peak of C=O stretching vibration moved to lower wave numbers with increasing soy protein. The reason could be ascribed to the formation of new hydrogen bonds between hydroxyl groups on PVA molecule and amide groups on SPI molecule, which enhanced the interaction between the protein and PVA phases and improved their compatibility.
X-ray Diffraction Analysis
The XRD curves of PVA/SPI blends were shown in Fig. 2. Glycerol plasticized soy protein showed a dominant amorphous structure exhibiting a broad arch centered at 20°. While the P10S0 showed an obvious diffraction peak at 19.8°, which was a characteristic crystallization peak of PVA. For the blends, diffraction peaks were found at about 2θ = 20°, indicating soy protein did not affect the crystalline structure of PVA. However, the intensity of the diffraction peak decreased with increasing SPI, indicating the decreasing crystallinity of PVA, which was a result of the interaction between soy protein moiety and PVA.

XRD patterns of PVA/SPI blend film
Differential Scanning Calorimetry Analysis
The melting and crystallization behaviors of PVA/SPI blend film were investigated by DSC and the thermograms were shown in Fig. 3. No melting peaks was observed on the exothermal curve of P0S10, indicating the amorphous nature of soy protein. For this reason, the endo- and/or exo-thermic peaks occurred on the thermograms were contributed by PVA component, such as the exothermic peak around 161 °C (Fig. 3). It was observed from the thermograms that the crystallization and melting temperatures (T c and T m ) decreased with increasing SPI content. Soy protein moiety could act as heterogeneous nucleation agent, resulting in the decreased crystallization temperature. The crystallization and melting enthalpy decreased with increasing protein content, indicating a decreased crystallinity of PVA. The strong hydrogen interaction between PVA and SPI may restrict the orderly compacting of PVA segments, leading to the decreased crystallinity.

The exothermic curves (a) and endothermic curves (b) of PVA/SPI blends
SEM
Figure 4 showed the scanning electron microscope (SEM) images of PVA/SPI blends with different content of SPI. Both neat PVA and neat soy protein films exhibited a smooth surface. All the blend film possessed a two-phase structure. The soy protein phase dispersed uniformly in the PVA matrix, implying a good adhesion between the two phases, resulting in the improved mechanical properties discussed below. Both soy protein and PVA possess polar groups on the backbones, resulting in strong hydrogen bonding interaction between them.

SEM photographs of PVA/SPI blend film with 0 (a), 10 (b), 20 (c), 30 (d), 40 (e) and 100 % (f) soy protein
Mechanical Properties
The mechanical properties of PVA/SPI blend film were shown in Fig. 5 and the typical stress–strain curves were shown in Fig. 6. Stress of the samples increased rapidly at small strains but very slowly after yielding. Glycerol plasticized neat PVA film exhibited a flexible character, with a tensile strength of 21 MPa and an elongation at break more than 1,600 %. Pure soy protein exhibited a brittle behavior, exhibiting low stress and strain at break. For the blends, with increasing soy protein, both tensile strength and elongation at break of the blends decreased, however, the Young’s modulus increased. For the strong hydrogen bonding between the two phases and the homogeneous distribution of protein phase in PVA matrix, the PVA/SPI blend film still possessed a good flexibility. When the SPI content increased to 30 %, the tensile strength maintained at 10 MPa and the elongation at break at 977.8 %, which was superior to the commonly used package film like LDPE.

Effect of SPI content on the mechanical properties of the blend film

Typical stress–stain curves of PVA/SPI blend film
Hydrophilicity
The water absorption behaviors of the blend film at a 58 % RH were shown in Fig. 7. For all the samples, the WU increased rapidly in the first 30 h and then reached a plateau, corresponding to absorption equilibrium. Figure 8 showed the WU at equilibrium and water diffusion coefficients as functions of soy protein content. Neat PVA film provided the lowest WU and the lowest diffusion coefficient. With increasing soy protein, both the WU and diffusion coefficient increased. The reason may be that the soy protein was more sensitive to water than PVA. In addition, as discussed above, the strong interactions inhibited the crystalline of PVA, resulting in the decreased crystallinity of blends, which increased the WU and diffusion coefficient, for water molecules could only transport through the amorphous region of the film.

Water uptake of PVA/SPI blend film as a function of time

Equilibrium water uptake and diffusion coefficient as a function of SPI contents conditioned at 58 % RH
TGA Analysis
The thermal stability was investigated using TGA, and the weight loss traces recorded in a range of 25–600 °C were shown in Fig. 9. For neat PVA film, the weight loss at around 100 °C was attributed to the evaporation of the absorbed water, and that around 220 °C was mainly related to the evaporation of glycerol plasticizer. The majority of mass loss took place at about 300 °C, followed by a further smaller mass loss at around 440 °C, which were attributed to the acetate group elimination at lower temperatures followed by a breakdown of polymer backbone at higher temperatures [25]. For neat SPI film, the weight loss between room temperature and 120 °C was attributed to the absorbed water and that in a temperature range of 120–250 °C was mainly related to the evaporation of glycerol. The degradation of protein mainly occurs at the temperature higher than 290 °C [26], and the residue weight was much higher than neat PVA, indicating the better thermostability of soy protein film. The PVA/SPI blend film exhibited similar water evaporation and glycerol release weight loss behaviors around 100 and 220 °C. However, the residue weight of PVA/SPI blend film after the thermo-degradation was higher than that of neat PVA, and increased with increasing soy protein. This behavior evidenced the improvement of thermostability caused by incorporation of SPI into PVA.

The TGA curves of PVA/SPI blend film
Conclusions
Poly vinyl alcohol/soy protein isolate (PVA/SPI) blend film plasticized with glycerol was fabricated by melting processing in presence of water. It was found that new hydrogen bonds between hydroxyl groups on PVA and amide groups on SPI were formed, which resulted in decrease of crystallinity of PVA, decrease in crystallization and melting temperatures. The lowered crystallinity in turn resulted in increase in both WU and diffusion rate. Although certain degree of phase separation occurred in the blend film, the soy protein moiety still dispersed uniformly in the PVA matrix and maintained a good compatibility between the two phases, which ensured the film retaining excellent mechanical properties. With 30 % protein content, a tensile strength of 10 MPa and an elongation at break higher than 970 % were observed. In addition, the incorporation of SPI rendered a higher thermostability to the PVA/SPI blend film than neat PVA.
References
Liu W, Li H, Wang X, Du Z, Zhang C (2013) Cell Polym 32:343
Reddy M, Mohanty AK, Misra M (2012) Macromol Mater Eng 297:455
Liu D, Wu Q, Chang PR, Gao G (2011) Carbohydr Polym 84:686
Tian H, Wu W, Guo G, Gaolun B, Jia Q, Xiang A (2012) J Food Eng 109:496
Zheng K, Zhang J, Cheng J (2013) Ind Eng Chem Res 52:14335
Dani J, Reddy J, Rajulu V (2011) Polym Comp 32:398
Sionkowska A (2011) Prog Polym Sci 36:1254
Song F, Tang D, Wang X, Wang Y (2011) Biomacromolecules 12:3369
Chen P, Zhang L (2005) Macromol Biosci 5:237
Liu D, Tian H (2012) Soy protein nanocomposites: properties and applications (Vol II, Chapter 21). In: John M (ed), Natural polymers. Royal Society of Chemistry
Liu D, Zhu C, Peng K, Guo Y, Chang PR, Cao X (2013) Ind Eng Chem Res 52:6177
Tian H, Zhang L (2010) Macromol Mater Eng 295:451
Tian H, Xu G (2011) J Polym Environ 19:582
Liu D, Tian H, Zhang L (2007) J Appl Polym Sci 106:130
Chen P, Zhang L, Cao F (2005) Macromol Biosci 5:872
Tian H, Zhang L (2010) Ind Crop Prod 32:13
Kumar R, Anandjiwala RD (2012) J Appl Polym Sci 124:3132
Wu W, Tian H, Xiang A (2012) J Polym Environ 20:63
Zhu Y, Du Z, Li H, Zhang C (2011) Polym Eng Sci 51:1770
Hameed N, Xiong R, Salim N, Guo Q (2013) Cellulose 20:2517
Su J, Yuan X, Huang Z, Xia W (2010) Polym Degrad Stab 95:1226
Su J, Huang Z, Zhao Y, Yuan X, Wang X, Li M (2010) Ind Crop Prod 31:266
Comyn J (1985) In: Comyn JE (ed) Polymer permeability. Elsevier Applied Science, New York
Vergnaud JM (1991) Liquid transport process in polymeric materials: modeling and industrial applications. Prentice-Hall, Englewood Cliffs
Holland BJ, Hay JN (2002) Polymer 43:2207
Chen P, Zhang L (2006) Biomacromolecules 7:1700
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Guo, G., Zhang, C., Du, Z. et al. Structure and Properties of Poly (Vinyl Alcohol)/Soy Protein Isolate Blend Film Fabricated Through Melt Processing. J Polym Environ 23, 183–189 (2015). https://doi.org/10.1007/s10924-014-0682-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-014-0682-7