
Abstract
The model polyurethane foam and model compact polyurethane material were prepared and then decomposed by means of natural oils. Castor oil and fish oil based polyol were used in this study. Optimal conditions for the polyurethane decomposition were found. Temperature 250 °C was necessary for efficient polyurethane decomposition by castor oil whereas 200 °C is sufficient in the case of fish oil based polyol. Prepared products have hydroxyl number in the range of 95–168 mg KOH g−1. During the polyurethane decomposition no cleavage of double bonds in the fatty acid chains of castor oil and fish oil based polyol was observed.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyurethane (PUR) scrap, especially of flexible foam, presents a serious problem of waste treatment but also a valuable source of materials. Therefore, many efforts have been done in the field of PUR recycling in order to find an optimal treatment for PUR waste. Various physical as well as chemical recycling methods were developed in the past. Physical (mechanical or material) recycling both of rigid and flexible PUR foams is today performed by injecting finely ground material into the stream of the components when producing new material with loads up to 15% by weight [1, 2]. Nevertheless, physical recycling of the PUR consumes at present about 120.000 t p.a. which is only 1% of the produced amount [3]. In the case of chemical recycling of PUR, the aim is to produce recycled polyols to be applied again in PUR formulations. Among the chemolysis methods of PUR, a glycolysis has been the most investigated [4–12]. The glycolysis involves heating the PUR scraps in the presence of glycols, mainly diethylene glycol (DEG) or dipropylene glycol (DPG), and catalysts performing transesterification reaction in order to obtain origin low molecular polyols [13]. In spite of numerous ways to produce polyols from PUR wastes, only a very limited number have been used in industrial scale. The reasons for this phenomenon are quality problems, economy of the process and the environmental balance of the process in comparison to the production of virgin polyols [3]. Due to these economical (the rising price of a crude oil) and environmental (the reduction of crude oil reserves) aspects, we have tried to develop new low-energetic recycling method, in which renewable agents instead of petrochemical glycols were used to decompose of PUR waste.
Nowadays, natural oil-based polyols (oleochemical polyols) are getting a lot of attention, and rising oil price has accelerated the polyurethane sector’s interest in such materials, as an alternative to current commercial polyols based on petroleum products [14–23]. From this point of view, vegetable oils and fats such as soybean oil, castor oil, palm oil, rapeseed oil, olive oil, and linseed oil are very important resources for polyols. Natural oils are triglycerides (triesters of glycerol with long-chain fatty acids) with varying composition of fatty acids [24]. For PUR chemistry, the most important oils are highly unsaturated oils, where, by using various chemical reactions, the double bonds are transformed into hydroxyl groups [14]. This requirement of highly unsaturated vegetable oils meets soybean oil, sunflower oil, safflower oil, corn oil, linseed oil, olive oil, tung oil, castor oil and others as well as an oil of animal origin: fish oil [14]. Usage of oleochemical polyols enables to prepare new PUR materials with beneficial properties coming from non-polar pendant chains of fatty acid [25]. In literature it is mentioned that PUR foams prepared from vegetable oil polyols exhibits improved hydrophobicity and thermal stability in comparison to those prepared from propylene oxide-based polyols [26].
Our idea was to use renewable resources (oleochemical polyols) in combination with recycled polyols for preparation of new PUR materials. To eliminate petrochemical polyols entirely, the oleochemical polyols were used as reagents for PUR decomposition instead of conventional petrochemical glycols. Surprisingly it was found that PUR decomposition via transesterification reactions of urethane and urea structures of PUR foam with hydroxyl group of the reagents takes place under conditions similar to classical glycolysis. A utilization of natural oils or their derivates as reagents for PUR chemolysis has not been mentioned yet in the literature. Krzan et al. only reported unsuccessful usage of castor oil for PET decomposition [27].
Therefore, the aim of our study was to evaluate natural oil-based polyols as possible new renewable reagents for chemical recycling of PUR waste. Model PUR materials were decomposed via transesterification reaction of urethane and urea structures with hydroxyl groups of the castor oil (CO) and the fish oil based polyol (FP) as deputies of vegetable and of animal resources, respectively. FP used in this study is produced by Icelandic Polyols Company (Icepol) from fish oil, which is a by-product of the fishing industry. Contrary to CO, which comprises secondary hydroxyl groups, FP contains mainly more reactive primary hydroxyl groups. Various reaction conditions for PUR decomposition were tested and monitored. The obtained products were analyzed by size exclusion chromatography (SEC), hydroxyl number, acid number and iodine number determinations.
Experimental
Model Polyurethane Materials—Preparation and Characterization
Materials
Polymeric methylene diphenyl 4,4′-diisocyanate (PMDI, Isotan 30) with NCO content = 7.5 mmol g−1 and trifunctional polypropylene ether polyol (Desmophen 5035 BT) with hydroxyl number = 36 mg KOH g−1 were kindly provided by Brahe (Czech Republic) and Bayer A.G. (Germany), respectively. Distilled water was used to prepare water-blown model PUR foam. Catalysts: 1,4-diazabicyclo[2.2.2]octane (DABCO) and dibutyltin dilaurate (DBTDL) were supplied by Aldrich (Germany). All materials were used as received.
Preparation
Model water-blown flexible PUR foam The formulation is given in Table 1. First, polyol, water and catalytic system were homogenized (120 s at 1,500 rpm) in plastic vessel. Then isocyanate was added and the system was quickly mixed (30 s at 1,800 rpm) and kept to free rise foaming. The prepared PUR foam was post-cured for 12 h at 55 °C.
Model compact PUR material The formulation is given in Table 1. All components were mixed together and after homogenization the mixture was placed into a closed glass mold to make sheet (about 3 mm thick), cured for 24 h at 25 °C and post cured for 24 h at elevated temperature (55 °C) to achieve full conversion of NCO groups. The prepared model PUR material contained no urea structures.
Characterization
Complete reactions were confirmed by ATR-FTIR test as disappearance of an isocyanate band at 2,270 cm−1. The ATR-FTIR spectra were collected at a resolution of 4 cm−1 and 16 scans per run, on the Perkin-Elmer Paragon 1000 PC. For each sample three measurements at different sample area were performed.
Thermal stability of the prepared model PUR foam and the compact material were characterized by thermogravimetric analysis (TGA) on the Perkin-Elmer 7 DSC with control and evaluation program Pyris 1. Around 3 mg of the sample was used for the analysis. Temperature dependences were measured at heating rate 10 °C min−1 in nitrogen atmosphere.
Decomposition of Model Polyurethane Materials
Materials
Castor oil (CO, puriss, Aldrich, Germany) and fish oil based polyol (FP, Icepol, Iceland) were used without further purification as reagents (decomposing agents) for decompositions of the prepared model PUR materials. The decompositions were carried out either with catalyst or without catalyst. Sodium hydroxide (NaOH, p.a., Lachner, Czech Republic) or diethanolamine (DEA, 98%, Aldrich, Germany) were used as catalysts without further purification. For comparison, a conventional glycolyses with dipropylene glycol (DPG, 98%, Aldrich, Germany) were also performed. Table 2 shows determined hydroxyl numbers (OHexper), acid numbers and iodine values (IVexper) of all reagents.
Decomposition
The decomposition of the prepared model PUR materials was performed in a 3-necked round bottom flask fitted with a stirrer, reflux condenser and nitrogen inlet. Decomposing reagent, chopped PUR materials and optionally catalyst (1 wt% of the PUR mass) were put into the flask and the reactor was then placed into salt bath preheated to the reaction temperature. All decompositions were performed at constant temperature (in the range of 180–250 °C) for 2 h. After the reaction, the reaction mixture was cooled to room temperature. The liquid product was filtered off by vacuum filtration separating it from solid residues and collected for further analysis.
Characterization
The reaction mixture was sampled at different reaction times and the progress of decomposition was evaluated by size exclusion chromatography (SEC). SEC was carried out with the Modular GPC System equipped with a refractive index detector RI 101 (Shodex, Japan) and UV–vis photometric detector UVD250 (Watrex, Czech Republic) operated at λ = 260 nm, and a set of two columns PLgel MIXED-C, 5 μm particle size, 300 mm × 7,5 mm (Polymer Laboratories, UK). Tetrahydrofuran (THF) was used as a mobile phase with a flow rate 1 cm3 min−1.
Hydroxyl number of the prepared products was experimentally determined by acetylating method according to ISO 2554:1974 (OHexper). Theoretical hydroxyl number (OHtheor) of the prepared products was calculated from known weights of the components (PUR, reagent and catalyst) of the reaction mixture and from experimentally determined hydroxyl number (OHexper) of the reagent. In the case that decomposition was catalyzed by DEA then OHexper of DEA (1,571 mg KOH g−1) was also taken into account. OHtheor of the product was then calculated according to the following equation:
where OHexper(reagent) and OHexper(DEA) were the determined hydroxyl numbers of the reagent and DEA, respectively, and m(reagent), m(DEA), m(PUR), m(catalyst) and m(solids) were weights of the reagent, DEA, the model flexible PUR foam, the catalyst and the solid residues after the decomposition, respectively.
Iodine value was determined by Hanus method based on the addition of iodine bromide to double bond of the fatty acid chain followed by titration of formed iodine by thiosulphate. Acid number determination was carried out according to ASTM D 4662-93.
Results and Discussion
Preparation of Model Polyurethanes
For the preparation of the model PUR foam, a suitable amount of water added into the formulation was chosen in order to create a chemical network where: 33 mol% of isocyanate groups of PMDI reacted with hydroxyl groups of polyether polyol resulting in formation of urethane (U) groups. Since stoichiometric ratio of isocyanate and hydroxyl groups was used (NCO index 100), it could be supposed that remaining 67 mol% of isocyanate groups of PMDI reacted with hydroxyl groups of water leading to a creation of mainly disubstituted urea (DU) structures. The molar ratio 1/2 of U/DU structures is common for commercial flexible PUR foams [28, 29]. On the contrary, the model compact PUR material was prepared without water addition in a sealed glass mold to prevent an access of air humidity. Thus, no DU structures were present in the material. From a known chemical composition of the model PUR materials, the content of U and DU structures were calculated. The calculation assumed equivalent weights of U and DU repeating units 1,686 and 230, respectively, taking into account the equivalent weights of the trifunctional polyol (Desmophen 5035 BT) and PMDI (Isotan 30) 1558 and 128, respectively. From known weights of Desmophen 5035 BT, Isotan 30 and water, it was calculated that the model PUR foam was composed of 78 wt% U repeating units and 22 wt% DU repeating units. Based on the above mentioned parameters, the model PUR foam contained 0.46 mmol g−1 U and 0.92 mmol g−1 DU structures and the compact PUR material contained only U structures in the concentration of 0.59 mmol g−1.
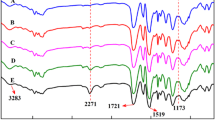
The both prepared fully-cured model PUR materials (foam and compact) comprised no isocyanate band (2,270 cm−1) in IR spectra after the post curing indicating complete polyaddition reaction (Fig. 1). The IR spectrum of the model compact PUR material showed up two bands characteristic for the U structure: at 1,727 and 1,537 cm−1 due to C=O stretching and N–H bending vibrations, respectively. In the IR spectrum of the model flexible PUR foam besides urethane absorption bands (1,727 and 1,537 cm−1) also bands at 1,662 and 1,510 cm−1 assigned to C=O stretching and N–H bending vibrations, respectively, of the DU structures originating from a reaction of added water with isocyanate were observed [30].

FTIR spectra of prepared model PUR materials: (i) flexible PUR foam and (ii) compact PUR material. Identification of bands: 1,727 cm−1 (C=O stretching vibration of urethane), 1,662 cm−1 (C=O stretching vibration of disubstituted urea), 1,537 cm−1 (N–H bending vibration of urethane) and 1,510 cm−1 (N–H bending vibration of disubstituted urea)
TGA curves (Fig. 2) showed good thermal stability of prepared PUR materials what was comparable to commercial flexible PUR foams. All tested PUR samples were thermally stable up to ca 250 °C. The compact PUR material containing only U structures exhibited narrow one-step degradation in nitrogen atmosphere. In the case of foamed PUR, from Fig. 2 broader degradation step was seen caused by the presence of various structures, mainly U and DU units. The amounts of solid residues after TGA around 6 wt% were observed in the cases of PUR foams whereas in the case of compact PUR material the solid residues present ca 1 wt%. Higher solid residue content corresponds to a higher content of thermally stable aromatic structures based on PMDI [31–34].

TGA curves (nitrogen atmosphere) of prepared (i) model PUR compact material and (ii) model flexible PUR foam, and (iii) commercial flexible PUR foam
Decomposition of Model Polyurethanes with Castor Oil
Reaction conditions suitable for a decomposition of PUR by glycols (glycolysis) were used as a starting point for a determination of optimal reaction conditions for PUR depolymerization by means of CO and RFO. Based on our previous research [5] and other published works [6–8, 11], the optimal conditions for glycolysis can be summarized as follows: reaction temperature: 190–230 °C, reaction time: 1–4 h, PUR foam/glycol weight ratio: 2 / 1–1 / 2, suitable catalyst: DEA or metal hydroxide.
Reaction conditions and results of decomposing experiments of model flexible PUR foam with dipropylene glycol (DPG), castor oil (CO) and their combinations are given in Table 3. The first experiments with DPG confirmed our previous results with waste PUR foams [5]. For a complete and fast PUR depolymerization the weight ratio PUR foam/reagent had to be 1/1, which means for the model PUR foam 10 times molar excess of hydroxyl groups per U and DU structures. A high excess of hydroxyl groups from the reagent favoured an equilibrium transurethane (or transurea) reaction between reagent and U (or DU) structure on the side of aliphatic and carbamate polyols formation (Scheme 1). No or low (run 1C) excess of reagent led to long reaction time and incomplete reaction. The model PUR foam decomposed easily also without addition of catalyst (run 3C). This fast and complete decomposition was catalyzed by catalytic system residues from the PUR foam formulation (the rest of DABCO and DBTDL), which were still present in the model PUR foam.

Decomposition reaction of model PUR foam based on 4.4′-diphenylmethane diisocyanate and polyetherpolyol with hydroxyl groups of oleochemical polyols
For PUR decomposition by CO it was necessary to increase reaction temperature up to 250 °C (see runs 4C–7C). Lower temperatures led to an incomplete PUR decomposition. A complete PUR foam dissolution with and without addition of the catalyst (DEA) was achieved after 25 and 26 min at 250 °C, respectively (runs 6C and 7C). The utilization of CO as reagent corresponded to higher PUR foam/reagent weight ratio in comparison with DPG due to a lower concentration of hydroxyl groups in CO. An effort to reduce the amount of reagent led to an incomplete PUR decomposition after 2 h of the reaction that was indicated by long dissolution of PUR foam and high amount of solid residues (9.5 wt%) in the liquid product (run 9C). The utilization of the PUR foam/CO weight ratio 1/1 (the same as for DPG) which meant twice molar excess of hydroxyl groups from CO against reaction stoichiometry [U + DU (foam)/OH (CO) molar ratio = 1/2], did not lead to complete PUR foam dissolution neither after 5.5 h of reaction. This resulted in a very high solid residue content, about 16.4 wt% (run 10C). During the PUR decomposition at 250 °C, also thermal degradation of the material could not be neglected, because the thermal decomposition of the prepared PUR foam started at temperature around 250 °C (Fig. 2). From this point of view, we tried to decrease the reaction temperature by utilization of various DPG and CO combinations (runs 11C–13C). The 25 and 50% molar replacements of hydroxyl groups of CO by hydroxyl groups of DPG did lead neither to faster nor to complete PUR foam dissolution. The liquid products with 10.5 and 13.4 wt% of the solid residues, respectively, were obtained. The 75% molar replacement of hydroxyl groups of CO by hydroxyl groups of DPG (run 13C) reduced solid residues (5.2 wt%), but still no complete foam dissolution after 2 h of reaction was achieved.
We could conclude that lower reactivity of CO comparing to DPG was caused by presence of only secondary hydroxyl groups in CO and their steric hindrance by long chain of ricinoleic acid in the molecule of triglyceride.
In Table 4 reaction conditions of decomposing experiments of the model compact PUR material with DPG, CO and their combination are given. Temperature 230 °C enabled to dissolve the model compact PUR in both reagents, in DPG as well as in CO. For this material, the PUR depolymerization by DPG was convenient at PUR/DPG weight ratio 1.7 / 1 corresponding to 15 times molar excess of OH groups from DPG to U structures in PUR material (runs 1BK and 2BK). Surprisingly, a use of CO enabled to carry out the PUR decomposition only with 5 times molar excess of OH groups. This was probably caused by a solubility phenomenon. In the beginning of the reaction, insoluble PUR crosslinked structure was decomposed to PUR oligomers, which started to go into solution in the reaction mixture. Then, the reaction continued with higher reaction rate in a homogeneous liquid phase. In the case of low amount of DPG in the reaction mixture, after ca 30 min the reaction led to the phase separation (an opalescence was observed) and to the formation of another solid insoluble phase. The reaction continued in heterogenic phase and therefore the reaction rate was significantly lowered.
From a comparison of the decomposition of the both model materials (the PUR foam and the compact PUR) it could be stated that the reaction of U structures with hydroxyl groups took place faster in comparison to DU structures.
Decomposition of Model Polyurethanes with Fish Oil-Based Polyol
Reaction conditions and results of decomposition of the model flexible PUR foam with FP are given in Table 5. In contrast to CO, FP contained primary hydroxyl groups which are generally more reactive than secondary ones. The decomposing experiments of the model PUR foam with FP confirmed this fact. As shown in Table 5 foam dissolution was very fast (5 min) at 230 °C (run 1CR). At 200 °C foam dissolution was also fast (ca 20 min) even if an excess of hydroxyl groups of FP was reduced to 5/1 molar ratio of hydroxyl groups (FP)/U and DU structures (PUR foam) without using of additional catalyst (runs 2CR and 3CR). Further temperature decreasing to 190 °C (run 4CR) led to slight increase of dissolution time (30 min) and the content of unreacted PUR solid residues to 1 wt%. At 180 °C (run 7CR), the dissolution of PUR foam was slower (60 min), nevertheless the content of the solid residues was still very low (0.4 wt%).
The previous mentioned experiments were conducted without any addition of catalyst. An addition of NaOH slightly reduced time of foam dissolution (runs 5CR and 6CR). The amount of solid residues in the liquid product after the PUR decomposition was very low (ca 0.1 wt%).
In Table 6 reaction conditions of decomposing experiments of the model compact PUR material with FP are given. After 2 h of decomposition no solid residues were present even at the lowest examined temperature, at 180 °C (run 4BKR). The molar excess of hydroxyl groups from FP is reduced to 5/1 molar ratio of OH groups/U structures in the model compact PUR. Under this ratio, a complete PUR dissolution under 20 min was observed in temperature range of 180–200 °C.
Characterization of Prepared Products
Products Prepared by Polyurethane Decomposition with Castor Oil
Size exclusion chromatography (SEC) was selected for characterization of the products prepared by complete decomposition of the model PUR foam (run 7C) and the model compact PUR material (run 3BK) by CO and the results are given in Figs. 3 and 4. In the RI SEC records, a peak with retention time of 16.0 min was assigned to original aliphatic polyol (Desmophen 5035 BT) from the model PUR foam (see the RI SEC record of the pure Desmophen 5035 BT also in Fig. 3) and another peak with retention time of 17.5 min corresponded with carbamate polyols (based on CO and PMDI) and unreacted CO. The aliphatic polyol had no UV signal (Fig. 4) which indicated complete decomposition of PUR network. No higher molecular weight structures (PUR oligomers) were observed in the SEC records. Strong UV signal of the peak of carbamate polyols indicated aromatic structures coming from polymeric MDI (Fig. 4).

SEC records (RI detector) of (i) the product obtained by decomposition of model PUR foam with castor oil (run 7C), (ii) the product obtained by decomposition of model PUR compact material with castor oil (run 3BK), (iii) pure castor oil and (iv) original aliphatic polyol from the model PUR foam

SEC records (UV detector) of (i) the product obtained by decomposition of model PUR foam with castor oil (run 7C), (ii) the product obtained by decomposition of model PUR compact material with castor oil (run 3BK), (iii) pure castor oil and (iv) original aliphatic polyol from the model PUR foam
SEC records (RI detector) of the products obtained by decompositions of the model PUR foam performed with addition of various catalysts are given in Fig. 5. It is clearly seen that the decompositions either with DEA catalyst (run 6C) or without catalyst (run 7C) led to the same product composition, which was indicated by similar SEC records. An addition of strong basic catalyst (NaOH) for the PUR decomposition led to a very viscous product which had physical gel-like character rather than liquid. The SEC record of this product contained the peak corresponding to the carbamate polyols and the unreacted CO (retention time 17.3 min), which was much broader than in the case of DEA-catalyzed or non-catalyzed systems. This indicated a presence of compounds with broader distribution of molecular weights, which were formed during the PUR decomposition by basic hydrolysis and transesterification reactions of the triglycerides of CO. The same phenomenon was also observed for PUR decomposition with FP catalyzed by NaOH. From this point of view, the weak bases (amines) seemed to be selective catalysts for the transurethane and the transurea reactions, whereas the strong bases (e.g. NaOH) promoted unwanted hydrolysis and transesterifications of ester bonds of the reagents (CO and FP), which led to a broad molecular distribution of the final products.

SEC records (RI detector) of the products obtained from model PUR foam decomposition with castor oil performed with different catalysts: (i) NaOH (run 8C), (ii) DEA (run 6C) and (iii) without catalyst (run 7C)
Products Prepared by Polyurethane Decomposition with Fish Oil-Based Polyol
As it was discussed before, the use of FP instead of CO enabled to increase the rates of PUR foam dissolution and the transurethane reaction. This fact caused that the depolymerization could be performed at lower temperatures than in the case of CO. Nevertheless, from Figs. 6 and 7 it is clearly seen that only the experiments performed at 200 °C led to a complete PUR network decomposition after 2 h of the reaction. During decomposing experiments at 180 °C, in the both cases with the model PUR foam and with the model compact PUR material, respectively, the polyols containing unreacted PUR structure units indicated by the peak at 15.9 min in UV SEC records were still present. The peak indicated aromatic structures from PMDI, which still remained bonded with aliphatic polyol units. Therefore in this case, the decomposition of the PUR material was not complete and longer reaction time was necessary for a complete PUR decomposition at 180 °C. No by-products were formed when the decomposition was performed at higher temperatures.

SEC records of the products obtained by decomposition of model PUR foam with fish oil-based polyol (i) at 200 °C (run 3CR) and (ii) at 180 °C (run 7CR) after 2 h of the reaction

SEC records of the products obtained by decomposition of model compact PUR material with fish oil-based polyol i) at 200 °C (run 2BKR) and ii) at 180 °C (run 4BKR) after 2 h of the reaction
In Fig. 8 RI SEC records of the completely decomposed products (runs 3CR and 2BKR), the pure FP and the aliphatic polyol (Demophen 5035 BT) are given. In comparison to the products based on CO, the SEC records of the products based on RFO contained three broad peaks with retention times 17.7, 18.2 and 18.9 min corresponding to the carbamate polyols and the unreacted FP. In the case of CO based product, the carbamate polyols were represented by one single peak in RI SEC record (see Fig. 3) due to “uniformity” of the unmodified CO containing only triglycerides, whereas in the case of FP based products, several types of the carbamate polyols were present originating from the mono-, di- and tri- acylesters of the FP. Therefore, the RI SEC record of the FP based product was more complex.

SEC records (RI detector) of (i) the product obtained by decomposition of model PUR foam with fish oil-based polyol (run 3CR), (ii) the product obtained by decomposition of model PUR compact material with fish oil-based polyol (run 2BKR), (iii) pure fish oil-based polyol and (iv) original aliphatic polyol from the model PUR foam
Further Product Characterization
The products obtained by the decompositions of both model PUR foam and compact PUR material by CO and FP were viscous liquids, yellow-orange and brown colored, respectively.
An amount of the aliphatic polyol (Desmophen 5035 BT) in the obtained products was determined from the RI SEC record as an area of the peak at retention time 16.0 min. The comparison of calculated (from known PUR compositions) and determined contents of the aliphatic polyol (Desmophen 5035 BT) in the products is given in Table 7. The values of experimentally determined contents of the aliphatic polyol were lower than calculated ones, which could be caused by different sensitivity of each component to RI detector.
From the view of further product application as polyols for PUR, the most important characteristic of the products are their hydroxyl numbers. From Table 8, it is seen no significant difference between calculated and experimentally determined hydroxyl numbers of the products. Generally, the products based on CO had lower contents of hydroxyl groups and the present hydroxyls were mainly secondary ones what meant less reactive. The products would be most suitable for applications where longer pot life is required (cast systems, coatings, etc.). On the other hand, the FP based products had higher hydroxyl number and due to the presence of primary hydroxyl groups they would react faster with isocyanates than the CO based products. Also no influence of reaction temperature (in the interval 230–250 °C) on the values of hydroxyl number was seen from the results (runs 4C–6C). The hydroxyl number slightly decreased with increasing reaction temperature (runs 2BKR, 3BKR and 4BKR), when the model compact PUR material was decomposed by FP. This decrease could be probably caused by certain loss of FP during the decomposition. At higher reaction temperatures, more volatile low molecular weight components of FP (e.g. alcohols) could be saturated into nitrogen flow leading to their escaping from the reactor. The content of the volatile compounds in FP was also confirmed by TGA (not shown here), when a weight loss during the heating from 30 to 200 °C was around 12%. A use of NaOH as catalyst slightly increased the value of hydroxyl number (run 8C), whereas DEA did not (run 7C).
In order to confirm that undesirable cleavage of double bonds of fatty acid chain during the PUR decomposition was not took place, iodine numbers of the products were determined. The comparison of calculated (theoretical) and experimentally determined values of the iodine numbers proved no cleavage of double bonds during the PUR decomposition (Table 8). This was valid for all performed experiments. From this point of view, the decomposition of PUR by means of CO and FP at temperature range 180–250 °C, either without or with catalysts (DEA, NaOH), did not lead to the cleavage of double bonds of the fatty acid chains. Acid numbers of the products are also given in Table 8. Slightly increased values of acid numbers were observed in the cases of products prepared at higher temperatures (230–250 °C).
Conclusions
The prepared model PUR materials, the flexible PUR foam as well as the compact PUR material, were successfully decomposed by natural oil-based polyols (CO and FP) and the liquid products with functional hydroxyl groups were obtained. The FP due to a content of primary hydroxyl groups reacted faster with U and DU structures than the CO and the PUR decomposition could be thus performed at lower temperatures. Hydroxyl groups of the both reagents (CO and FP) reacted slower with DU structures than with U structures of PUR. The prepared products had hydroxyl numbers in the range of 95–168 mg KOH g−1. During the PUR decomposition no cleavage of double bonds of the fatty acid chains of the CO and the FP was observed.
References
Stone H (1998) In: Frisch KC, Klempner D, Prentice G (eds) Advanced in plastic recycling: recycling of polyurethanes, vol 1. Technomic Publishing Company, Lancester, pp 167–222
Stone H, Villwock R, Martel B (2000) Polyurethanes conference. API, Boston
Behrendt G, Naber BW (2009) J University Chem Technol Metall 44:3
Nikje MMA, Nikrah M, Aga Mohammadi FH (2008) J Cell Plast 44:367
Benes H, Rosner J, Holler P, Synkova H, Kotek J, Horak Z (2007) Polym Adv Technol 18:149
Molero C, de Lucas A, Rodriguez JF (2006) Polym Degrad Stab 91:894
Molero C, de Lucas A, Rodriguez JF (2008) Polym Degrad Stab 93:353
Molero C, de Lucas A, Rodriguez JF (2006) Polym Degrad Stab 91:221
Datta J, Rohn M (2008) Polimery 53:871
Datta J, Pniewska K (2008) Polimery 53:27
Molero C, de Lucas A, Rodriguez JF (2008) J Appl Polym Sci 109:617
Nikje MMA, Garmarudi AB, Haghshenas M (2007) Polym-Plast Technol Eng 46:265
Scheirs J (1998) Polymer recycling. Wiley, New York
Ionescu M (2005) Chemistry and technology of polyols for polyurethanes. Rapra, Shawbury
Sharma V, Kundu PP (2006) Prog Polym Sci 31:983
Yeganeh H, Hojati-Talemi P (2007) Polym Degrad Stab 92:480
Zlatanic A, Lava CH, Zhang W, Petrovic ZS (2004) J Polym Sci, Part B: Polym Phys 42:809
Guo A, Demydov D, Zhang W, Petrovic ZS (2002) J Polym Environ 10:49
Ferrer MC, Babb D, Ryan AJ (2008) Polymer 49:3279
Kong X, Narine SS (2007) Biomacromolecules 8:2203
John J, Bhattacharya M, Turner RB (2002) J Appl Polym Sci 86:3097
Javni I, Zhang W, Petrovic ZS (2004) J Polym Environ 12:123
Petrovic ZS, Cvetkovic I, Hong DP, Wan X, Zhang W, Abraham T, Malsam J (2008) J Appl Polym Sci 108:1184
Gunstone F (1996) Fatty acid and lipid chemistry. Blackie Academic, New York
Raquez JM, Deleglise M, Lacrampe MF, Krawczak P (2010) Prog Polym Sci 35:487
Guo A, Javni I, Petrovic Z (2000) J Appl Polym Sci 77:467
Krzan A (1999) Polym Adv Technol 10:603
Verhelst G, Parfondry A (2002) In: Randall D, Lee S (eds) The polyurethanes book. Wiley, UK, pp 169–188
Bailey FE Jr (1991) In: Klempner D, Frisch KC (eds) Handbook of polymeric foams and foam technology. Hanser, Munich, pp 47–72
David DJ, Staley HB (1979) Analytical chemistry of the polyurethanes, part III. Robert E. Krieger Publishing, Huntington, New York
Rek V, Hace D, Bravar M, Jagodar A (1990) Angew Macromol Chem 176:135
Backus JK, Bernard DL, Darr WC, Saunders JH (1968) J Appl Polym Sci 12:1053
Herrera M, Wilhelm M, Matuschek G, Kettrup A (2000) J Anal Appl Pyrolysis 58:173
Ravey M, Pearce EM (1997) J Appl Polym Sci 63:47
Acknowledgments
The authors are grateful to the Ministry of Industry and Trade of the Czech Republic (grant no. 2A-2TP1/135) for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Beneš, H., Černá, R., Ďuračková, A. et al. Utilization of Natural Oils for Decomposition of Polyurethanes. J Polym Environ 20, 175–185 (2012). https://doi.org/10.1007/s10924-011-0339-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10924-011-0339-8