
Abstract
Cell motility makes essential contributions to normal embryonic development and homeostasis. It is also thought to contribute in important ways to tumor metastasis. Because of this dual importance, cell migration has been extensively studied. The fruit fly Drosophila melanogaster has served as an important model organism for genetic analysis of many aspects of developmental biology, including cell migration. Here we describe the various types of cell movements that have been studied in detail, which represent models for epithelial-to-mesenchymal transition, transepithelial migration, inflammation, wound healing and invasion. We summarize what has been learned about the molecular control of cell migration from genetic studies in the fly. In addition, we describe recent efforts to model tumor metastasis directly in Drosophila by expressing oncogenes and/or mutating tumor suppressor genes. Together these studies suggest that Drosophila has much to offer as a model for varied aspects of tumor metastasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cancer is a complex collection of diseases that result from misregulation of multiple biological processes, including cell division, cell migration, and apoptosis. However, the toughest battle in cancer treatment is to prevent and treat metastatic disease, which is the major cause of death for cancer patients. As metastatic cells invade surrounding tissues, they interact in complex ways with other cells in the microenvironment, with extracellular matrix, and with secreted molecules such as growth factors and cytokines. Moreover, the intrusive tumor cells have to survive attack from the immune system and adapt to new environments [1]. Metastasis is a process that is believed to require activation of a transcriptional program that alters cell–cell and cell-matrix adhesion as well as the cytoskeleton and thereby stimulates survival and migration. Aside from the contribution that abnormal cell migration makes to metastasis, cell migration is also a striking feature of normal animal development and is a subject of much investigation in this context. These two kinds of cell movements share common features. Therefore, a detailed understanding of the basic mechanisms of cell motility in model systems is likely to facilitate understanding—and treatment—of human metastatic disease.
Overview of Cell Migration in Drosophila
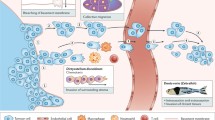
Drosophila is a popular model organism for the study of many features of embryonic development, most notably pattern formation. Important insights into basic developmental biology have emerged from this model organism, some of which have direct cancer relevance. For example, Hedgehog was first discovered based on its function in pattern formation of the Drosophila embryo [2]. Now it is known that the mammalian homologs of this secreted factor are critically important in normal human development and an increasing number of human cancers [3]. Such findings have led to the general idea that studies in model organisms such as flies have much to offer cancer biology. Given the importance of cell migration to normal development and tumor metastasis, investigators have sought to describe and understand the many stereotyped cell migrations that occur during Drosophila development. Each type has provided different information about the genetic control of cell motility and the mechanisms controlling the guidance of migrating cells to their targets (Table 1). Here we review briefly most of the embryonic cell migrations that have been studied in detail (Fig. 1), before focusing in more depth on two adult models: border cell migration in the ovary and recent efforts to model tumor metastasis by over-expressing oncogenes and mutating tumor suppressors in the fly (Fig. 2).

Drosophila models of cell migration. a–d Schematic drawings of mesoderm invagination in the Drosophila embryo. a Lateral view. b–d Cross-sections of the slice shown in a at increasing stages of maturity, dorsal is up. b Mesoderm cell fates (red) are specified at the ventral surface under the control of the twist and snail genes. c Invagination of mesodermal cells occurs and expression of FGF-like ligands initiates in ectodermal cells shown in blue. d FGF-like ligands thisbe and pyramus start to have distinct expression patterns. Mesodermal cells spread out over the ectoderm and migrate dorsally under the control of FGF-like ligands acting on an FGF receptor known as Heartless expressed in the mesoderm. e–f Schematic drawings of transepithelial migration of the primordial germ cells (yellow) through the midgut in Drosophila. e Lateral view, anterior is to the left, dorsal is up. f, g Magnified view of region boxed in e. Germ cells cross the posterior midgut epithelium (white) and then are repelled by the tissue shown in red and attracted by the somatic mesoderm shown in bright green. h–l) Schematic drawings of branching morphogenesis and tube formation during tracheal development. h A small patch of ectoderm on the surface of the embryo is shown in white. Cells expressing the FGF-like protein Branchless are interior to the ectoderm, and shown in blue. Breathless, an FGF-like receptor, is expressed in a subset of ectodermal cells (red). i Cells in which Breathless is activated invaginate to form multicellular tubes, shown by the dotted line in cross-section. j The pattern of Branchless expression (blue cells) changes as branching proceeds and finer, unicellular branches are formed, also shown in cross-sections. k Fusion of branches takes place between repeating segments of tubes to generate an interconnected system of branches by the end of embryogenesis. l After the large branches fuse, smaller tubes continue to branch off, also shown in cross-section. m–p Schematic drawing of border cell migration. m A whole adult fly showing the disposition of the ovary (white) within the abdomen and a single ovariole (red) within the ovary. n–p Higher magnification view of part of the ovariole indicated in m. Egg chambers of increasing stages of maturity are arranged in a row. Egg chambers are composed of germ cells (white) surrounded by epithelial follicle cells (gray) and a muscular sheath (red). n A pair of polar cells (yellow) at the anterior end of the egg chamber secretes a cytokine, which activates the JAK/STAT pathway in immediately adjacent cells to specify the border cells (blue). o Border cells detach from the epithelium and migrate in between the germ-line cells during stage 9 and arrive at the nurse cell/oocyte border at stage 10 (p). q At higher magnification, border cells show high levels of Drosophila E-cadherin (red) between the cells of the cluster and the polar cells.

Two models of tumor metastasis in fruit flies. a Model 1. Cells are taken from a larva that is homozygous for a tumor-suppressor gene such as lgl, dlg, or brat, and also carrying a permanent genetic marker, such as lacZ, and are used as donor tissue. This tissue is dissected out and injected into the abdomen of an adult fly. After a period of time, the fly is examined for lacZ-expressing cells at ectopic sites (black arrows) from the injection site (purple arrow), indicating metastatic behavior. In some cases, metastases are found inside the muscle sheath of the ovariole, (see Fig. 1m). To test for suppressors of tumorigenesis and metastasis, screens have been carried out using larval donor tissue homozygous for a second mutation in addition to the tumor-suppressor. b Model 2. Create marked tumors as clones of cells. In these studies, a larva carries a stable transgene encoding activated Ras (RasV12) and a GFP marker, but both are repressed by the Gal80 repressor. They may also bear additional heterozygous mutations, such as scribble; this mutation and the repressor are both distal to a recombination site. Recombination occurs in a few cells upon tissue-specific expression of a yeast recombinase, resulting in loss of the repressor in these cells and homozygosis of the other mutation, if present. Cells with activated Ras will overgrow as a clone of cells to create a tumor. After a period of time, larvae are examined to determine if GFP-positive tumor cells (shown by the green ovals with red outline) are located at ectopic sites.
Epithelial to Mesenchymal Transition (EMT) and Transepithelial Migration in Drosophila
During gastrulation in the fly embryo, cells on the ventral surface invaginate and then spread along the ectoderm in response to the transcription factors Twist and Snail (Fig. 1a–d) [4]. The orthologs of these proteins, Twist and Snail/Slug, are now thought to control cell migration more generally, both in vertebrate development and in the metastasis of many tumor types [5]. Specifically these transcription factors have been implicated in the process known as epithelial to mesenchymal transition (EMT), which is one mechanism by which cells that start out as part of an epithelium can acquire motile and invasive properties [6]. The transcriptional program initiated by Twist and Snail/Slug results in downregulation of cell–cell adhesion and conversion of polarized epithelial cells into invasive mesenchymal cells with an amorphous, fibroblast-like morphology [7]. This is an important mechanism that operates both during embryonic development and in tumor metastasis; however, as described later in this review, it is not always necessary for epithelial cells to undergo a complete EMT in order to escape from an epithelium, and ovarian epithelial cells have adopted another strategy. Immediately after gastrulation in the fly, the newly invaginated mesodermal cells spread out and move dorsally in response to two partially redundant FGF-like molecules acting on an FGF receptor [8, 9].
In a very different form of movement, the primordial germ cells undergo a transepithelial migration at mid-embryogenesis, traversing the midgut epithelium in a process similar to the extravasation of immune or tumor cells in mammals (Fig. 1e–g). Drosophila germ cells require activation of a CXCR4-like gene, tre1, to execute this movement [10]. Subsequently they are guided to the somatic gonad by a mechanism in which the migrating germ cells compete with neighboring somatic tissues for a lipid chemoattractant and survival factor [11].
Branching Morphogenesis and Tube Formation
Although there are no blood or lymphatic vessels in Drosophila—instead there is a tube-like heart and an open circulatory system—the development of the tracheal system a bit later in embryogenesis represents a model for angiogenesis (Fig. 1h–l). About 80 cells in each half-segment organize themselves into tubes [12]. Cells at the tips of the tubes grow out into peripheral tissues, forming finer and finer branches as they ramify along stereotyped trajectories. A secreted FGF-like protein, known as Branchless, plays a major role in patterning the branches and guiding their outgrowth. Branchless binds and activates a receptor tyrosine kinase known as Breathless. Many molecular details of the regulation of this complex process have been uncovered but will not be reviewed here due to space limitations [13, 14].
Models of Immunity, Inflammation, and Wound Healing
Despite the absence of blood vessels, circulating macrophages called hemocytes serve some of the functions of mammalian blood cells, specifically lymphocytes. Hemocytes depend on signaling through the single fly homolog of the PDGF and VEGF receptors (PVR) for their survival [15]. PVR activity is also required for the stereotyped movements of the hemocytes during embryogenesis [16, 17]. Thus, other molecules implicated in angiogenesis may also be conserved in these fly blood cells, and it is currently an emerging field of study.
Drosophila models of inflammation and wound healing have also been described. For example, lasers can be used to damage the epidermal layer of the embryo. These wounds heal rapidly, and as they do, hemocytes migrate to the wound site and engulf debris and bacteria [18]. In contrast to the requirement for PVR in the developmental migrations of hemocytes, PVR does not mediate the attraction of hemocytes to wounds [17], and it is unknown what does. A normal developmental process with some morphological similarity to wound healing is that of dorsal closure, in which the epithelial sheet of epidermis that encloses the ventral and lateral parts of the embryo “zips” together to cover the dorsal side of the embryo. Cells at the leading edge crawl up and over the dorsal tissue. Dorsal closure relies on Jun N-terminal Kinase (JNK) signaling, though it is as yet unclear precisely how dorsal closure is initiated or what activates the JNK pathway [19].
Thus, Drosophila embryos offer models for a variety of types of cell motility that are potentially relevant to tumor metastasis: EMT, trans-epithelial migration, inflammation, wound healing, and angiogenesis. These processes resemble aspects of metastasis not only at the cellular, descriptive level but also at the molecular level, as receptor tyrosine kinases such as FGF receptors and PVR relay important proliferation, survival and migration signals to the migrating cells.
Modeling Metastasis in Mature Flies
In addition to these embryonic models, two types of metastasis models have been developed in later stages of the fly lifecycle. In the adult ovary, a cluster of somatic cells known as the border cells undergoes an obligatory invasive migration as part of their normal development (see Fig. 1m–p). These cells start out fully integrated into the follicle cell epithelium. At a well-defined stage in development, they begin extending protrusions in between the cells of adjacent germ-line tissue. Ultimately they detach from the epithelium and migrate, as a coherent cluster, 150–200 μm between germ-line cells over the course of 4–6 h. If the cells fail to migrate, female sterility results, as their functions are essential. One of the major advantages of border cell migration as a model is the precision with which cell–cell interactions can be observed and manipulated. As a result, a detailed understanding has developed regarding the molecular and cellular interactions that govern their behavior.
A second adult model of migration and metastasis that will be described is the recent effort to model metastasis directly by expressing oncogenes, such as activated Ras, and mutating tumor suppressors. Remarkably, these genetic manipulations lead to tissue overgrowth and then spreading of the “tumor” cells throughout the body. In addition, examination of growth regulation in the fly reveals many analogies to tumorigenesis in mammals [20]. While a comprehensive explanation of tumor-suppression in the fly is beyond the scope of this review, we will discuss the recent efforts to model metastasis.
Border Cell Migration
The ever-expanding set of genetic tools available in fruit flies has been used to dissect the molecular mechanisms that control the interactions between border cells and their microenvironment. Many important genes have been identified, and remarkably, most have been linked to tumor metastasis [21]. Border cells are derived from the epithelial layer of the Drosophila egg chamber, which is composed of approximately 650 follicle cells in a monolayer that covers the outside of the germline cyst, itself composed of 15 nurse cells and one oocyte (Fig. 1m–q). Two specialized follicle cells, known as polar cells, located in the center of border cell cluster, secrete the ligand, Unpaired (UPD). UPD promotes the recruitment of four to eight neighboring follicle cells to become motile border cells by activating JAK/STAT signaling [22, 23]. Activated STAT continues to be required for efficient movement of the border cell cluster throughout its four to six hour journey [22]. Slow border cells (slbo), which encodes a basic region/leucine zipper C/EBP transcription factor, is one of the genes activated by JAK/STAT signaling and in turn activates downstream genes, including focal adhesion kinase (FAK), singed (sn, fly fascin), Drosophila E-cadherin (DE-cad), Armadillo (ARM, Drosophila β-catenin), and others, to regulate cell migration [24–27]. Of course, mammalian STAT proteins are important in normal breast development and have been implicated in breast cancer as well.
Once formed, the border cell cluster detaches from the anterior epithelium and is guided to the oocyte by the secreted ligands for the EGF receptor [28–30] and PVF1 (PDGF- and VEGF-related Factor 1). In flies there are four ligands that can activate the EGFR: Gurken, Keren, Spitz, and Vein. Two of them, Keren and Spitz, are expressed in the oocyte at stage 9 and have the ability to redirect border cells, when expressed ectopically [28]. PVF1, which is a secreted protein that activates a receptor tyrosine kinase known as PVR, is also sufficient to attract the moving border cell cluster to ectopic sites of expression. PVR and EGFR function redundantly, and both receptors must be downregulated to observe a strong defect in movement of the border cells to the oocyte. The TGF-alpha-like ligand, Gurken, is required near the end of border cell migration, when they get close to the oocyte. This ligand is expressed in a highly localized dorsal/anterior crescent in the oocyte and causes the border cells to veer off their central path and move to the dorsal side just as they reach the oocyte border.
Mammalian homologs of EGFR (ErbB1/HER1, ErbB2/HER2, ErbB3/HER3 and ErbB4/HER4) have been extensively studied in cancer, especially breast cancer. For example, HER2/ErbB2 is commonly amplified in breast cancers, and such cancers are generally more aggressive and have historically correlated with a poorer prognosis [31]. Moreover, there is ample evidence that many of the mammalian EGFR family members mediate migration and chemotaxis and that this contributes to their effects on cancer cells [32, 33].
The PEA3 group of the ETS family of transcription factors is also implicated in promoting metastatic behavior [34]. One fly ETS protein, YAN/AOP, is required for border cell migration as well as for the movements of epithelial sheets during dorsal closure. In border cells, Yan is regulated by several of the key signaling pathways that affect their migration. Yan expression turns on in response to STAT signaling just as the cluster becomes motile, but Yan protein levels are down-regulated during migration in response to increasing receptor tyrosine kinase signaling via PVR and EGFR as the cells move. yan mutations result in up-regulation of DE-cadherin, possibly due to alterations in endocytosis [35].
E-Cadherin in Border Cell Migration and Metastasis
Surprisingly, during border cell migration DE-cadherin and ARM are highly expressed [36]. This seems counter-intuitive since E-cadherin expression is reduced in many migrating cells. One explanation is that border cells migrate directly over the surfaces of the nurse cells, rather than through an extracellular matrix. Therefore, they may use DE-cadherin mediated adhesion much the way other migrating cells employ integrin. In addition, the details of E-cadherin localization in border cells are relevant: DE-cadherin accumulates to highest levels at the interface between the central non-migratory cells (polar cells) and the migratory cells (Fig. 1q). High levels of DE-cadherin are also found in adherens junctions that persist between the cells even as they migrate [37]. In fact, border cells retain apical/basal polarity throughout their migration [36, 38]. The lowest levels of DE-cadherin are found at the interface between migrating border cells and their substrate, the nurse cells (see Fig. 1q). The small amount of protein that is found there is functionally important because loss of DE-cadherin only from the nurse cells impedes border cell migration [36].
DE-cadherin serves multiple functions during border cell migration. DE-cadherin-mediated adhesion between border cells and nurse cells is thought to provide the traction necessary for border cells to move forward [37, 39]. DE-cadherin between border cells is believed to hold them together, and DE-cadherin between border cells and polar cells ensures that the migratory cells do not detach from the group. This mechanism is important because the migrating cells require continuous JAK/STAT signaling, which depends upon UPD secreted by the polar cells, in order to sustain their motility [22]. DE-cadherin expression levels must be regulated carefully within the border cells because loss of DE-cadherin from the cell cluster [36], or over-expression of DE-cadherin [35], blocks their migration. Therefore, although migrating border cells express higher levels of E-cadherin than the rest of the follicle cell epithelium, the regulation of DE-cadherin expression level, localization, and activity are clearly critical to achieve the adhesion necessary for traction and de-adhesion required for cells to retract the trailing edge and move forward.
Downregulation of E-cadherin in cancer cells is generally thought to promote metastatic behavior. For example, loss of E-cadherin is considered a marker for invasive lobular breast cancer (ILBC). More than 85% of ILBC patients display irreversible loss of E-cadherin that results from loss of heterozygosity at the CDH1 locus [40–42]. Nevertheless, the role of E-cadherin is not simply as a tumor suppressor gene. In breast cancer, E-cadherin is lost in most ILBC but in other subtypes it remains expressed [42]. In other cancers, such as ovarian cancer, E-cadherin levels are typically higher in the invasive carcinoma than in the normal surface epithelium. Border cell migration may therefore serve as an instructive model for understanding how tumor cells that maintain relatively high levels of E-cadherin still manage to migrate and metastasize.
In keeping with this idea, Myosin VI, an actin motor protein, was found to be important for the regulation of DE-cadherin levels in border cells and to be over-expressed in ovarian and prostate carcinoma. As mentioned above, achieving the appropriate level of DE-cadherin expression in border cells is essential. One factor that is required to stabilize DE-cadherin in border cells is the unconventional motor protein Myosin VI [43]. Interestingly human Myosin VI is highly expressed in the most aggressive forms of ovarian cancer, and stably down-regulation of this expression impedes their migration in vitro and their ability to metastasize in a mouse xenograft model [44]. Myosin VI expression is also upregulated in prostate cancer, and was selected for further study because of the link to border cell migration and ovarian carcinoma [45]. It will be interesting to see how generally Myosin VI turns out to contribute to carcinoma invasion and metastasis.
Steroid Hormones in Developmental Transitions and Border Cell Migration
In addition to the cytokine and growth factor signals that control border cell migration, the steroid hormone ecdysone plays a critical role. Ecdysone is the only known insect steroid hormone and exerts its biological effects by binding to a heterodimeric receptor composed of a ligand binding subunit, EcR (Ecdysone Receptor), and the RXR homolog, USP (Ultraspiracle). Ecdysone is best-known for its functions in developmental transitions, such as larval molting and metamorphosis. At first glance, these processes would seem to have little in common with human growth and development. However, it is interesting to note that metamorphosis, like puberty, is a transition from a sexually immature juvenile to the mature adult. Puberty and metamorphosis both depend critically upon increases in the concentration of circulating steroid hormones. Another similarity is that, in the fly as in humans, ovarian development depends upon steroid hormone action. Ecdysone levels are low early in egg chamber development. At stage 9, the same stage at which the border cells migrate, ecdysone levels rise precipitously and remain high through stage 10. Mutations that prevent ecdysone synthesis result in developmental arrest at stage 8. Similarly, global loss of ecdysone receptor function results in developmental arrest [46]. However, if ecdysone receptor function is compromised specifically in the border cells, the rest of ovarian development proceeds normally but border cells fail to migrate [37, 47]. In addition, an ecdysone receptor coactivator similar to AIB1 (amplified in breast cancer 1) was identified in a genetic screen for mutations that disrupt border cell migration. The Drosophila P160 coactivator is called Taiman (TAI), which means “too slow” in Chinese to describe its loss-of-function phenotype. TAI is a member of the p160 family of steroid hormone receptor coactivators. In mice and humans there are three P160 proteins, SRC1, SRC2, and SRC3, which is another name for AIB1.
AIB1 is frequently amplified or over-expressed in breast and ovarian cancers. It is a bona fide breast cancer oncogene because over-expression of AIB1 in mouse mammary tissue is sufficient to cause mammary hyperplasia and increases the frequency of mammary tumors [48]. Knock-out of AIB1 also reduces the incidence and growth of mammary tumors in mice [49]. AIB1 and other members of the family bind to steroid hormone receptors in a ligand-dependent manner and recruit additional transcriptional machinery. Likewise, in Drosophila, TAI binds to EcR/USP in a ligand-dependent manner and potentiates transcriptional activation. Therefore understanding more about the function of TAI may provide insight into the function of AIB1. Consistent with this idea, a recent study explored the idea that AIB1 might contribute to cell motility in human cancer cells as it does in border cells. Yoshida et al. showed that AIB1 was over-expressed in the most aggressive forms of ovarian cancer and that reduction in AIB1 expression inhibited the migration of ovarian carcinoma cells in vitro [50], a biological effect that had not previously been associated with AIB1. While the effects of AIB1 on both breast and ovarian cancer cells can be independent of estrogen receptor [50, 51], it remains to be seen if another hormone receptor is involved or if AIB1 is acting as a coactivator for a hormone-independent transcription factor. In border cell migration, it is clear that ecdysone signaling is required together with TAI; however, there is also evidence for ecdysone-independent transcription of downstream target genes in these cells (Starz-Gaiano and Montell, unpublished results).
One mechanism by which ecdysone, EcR and TAI might regulate border cell migration is by affecting cell adhesion. Border cell clones mutant for tai have elevated DE-cadherin accumulation between border cells and nurse cells [37]. This phenomenon may contribute to the migration defect because simply overexpressing DE-cadherin in border cells is sufficient to impede migration [35], however there are likely many additional effects. The regulation of cell adhesion is an important but incompletely understood feature of both border cells and metastatic tumor cells and is a subject that requires further investigation.
Knock-out mice have been generated which lack each of the three P160 coactivator family members. However functional analysis in vivo is challenging because of possible redundancy amongst the three genes and pleiotropies, such as decreased growth hormone and estrogen production. These effects, while physiologically important, make it difficult to sort out the direct and cell autonomous effects in particular cell types. The presence of a single protein in the fly, together with the ability to carry out mosaic analysis and study a specific biological readout (border cell migration) may lead to important insights into P160 coactivator function in vivo, that would be harder to determine in the mammalian system.
An Emerging Role for Notch Signaling in Cell Migration
Although a multitude of functions have been attributed to Notch both in normal development and in cancer [52], its ability to promote cell migration has only relatively recently been recognized. Notch has now been suggested to function in EMT [7] and a variety of other types of cell migration, including in Drosophila migration of glial cells [53] and border cells [26, 35, 54, 55]. Although Notch and its ligands are uniformly expressed in Drosophila egg chambers, Notch is specifically activated in border cells during their migration, due at least in part to elevated expression of the metalloproteinase Kuzbanian at that stage of development [26, 55]. This effect was demonstrated to be a relatively direct effect on migration rather than an indirect consequence of a defect in cell differentiation [55]. It is unclear precisely what aspect of border cell migration is regulated by Notch; however, the observation that constitutively active Notch does not impede migration is distinctive. All of the other signaling pathways known to affect border cell migration actually cause migration defects when they are hyperactivated. For example, over-expression of PVF1 or ligands for the EGF receptor impedes border cell migration. Similarly, hyperactivation of the STAT pathway can lead to migration defects. In contrast, constitutively active Notch is compatible with normal migration. Mouse mammary tumors frequently have a retroviral insertion into the Notch4 gene that results in expression of a truncated, constitutively active form of the protein [56]. This observation is interesting because it suggests that constitutive activation of the Notch family might promote cell motility in breast cancer as it does in border cells, in addition to the well-documented effects of Notch signaling on proliferation and survival.
Modeling Metastasis in Drosophila
In addition to studying normal, developmentally regulated cell migration, some researchers are taking a more direct approach to modeling metastasis in the fly. Several tumor suppressor genes, including lethal (2) giant larvae (lgl), discs large (dlg) and scribble (scrib), had long been known to promote tissue overgrowth when disrupted during fly development. These proteins are widely expressed and normally act together to regulate apical-basal polarity in epithelial cells, although additional functions are becoming more evident [57]. They are inter-dependent for proper sub-cellular localization in the plasma membrane. SCRIB and DLG localize to septate junctions, which although functionally analogous to tight junctions in mammalian cells, are located just basal to the adherens junctions, in the lateral membranes of epithelial cells. LGL is both cytoplasmic and found on baso-lateral membranes. The mechanism by which these proteins control polarity is not entirely clear at the molecular level, but they are proposed to work as scaffolding proteins that control the segregation of membrane proteins to distinct domains of the cell. In addition, they affect downstream signaling events, such as the activation of Rho GTPases. SCRIB has four PDZ domains and a stretch of leucine-rich repeats. DLG has three PDZ domains and is a membrane-associated guanylate kinase (MAGUK). LGL contains WD40 repeats. All of these genes have highly conserved homologs: there is one human Scribble, also called Vartul (although there are other possible functional orthologs), two LGL homologs (Lgl or Hugl-1, 2), and four DLG homologs. Interestingly, hScrib and Lgl-1 can functionally substitute for their fly counterparts in the fly [58, 59]. hScrib, Dlg1, and Dlg4 are targets for ubiquitin-mediated degradation by human papillomavirus E6 [60], and recent evidence is beginning to suggest they have a role in cell cycle progression of mammalian cells [61]. For example, overexpression of hScrib in some cultured human and mouse cell lines blocks the cell cycle at the G1/S transition and results in upregulation of APC (Adenomatous Polyposis Coli) and downregulation of Cyclins A and D, while knock-down of hScrib by siRNA promotes progression from G1 to S phase [62]. In addition, mice homozygous mutant for Dlg die early in embryogenesis and have overproliferation defects in the lens epithelium similar to those caused by expression of E6 [63].
Drosophila SCRIB, LGL and DLG are required in ovarian follicle cells to maintain organization of the epithelium. Loss of dlg, for example, results in a dramatic invasion of the epithelium into the nurse cells [64]. This invasive phenotype is dependent on Bazooka [65], the fly Par-3, a protein that is part of a multiprotein complex necessary for organizing the apical domains of epithelial cells. Since DLG, SCRIB, and LGL generally antagonize the function of polarity proteins in the Bazooka/Par6/aPKC complex, the invasion phenotype may be a consequence of loss of organized polarity. This apical complex is also required for organization of the border cell cluster and for efficient movement [38]. Other baso-lateral complex proteins are continuing to be identified in the fly, including fasciclin II (fas2). Overexpression of wild-type fas2 in border cells blocks their movement while fas2 with PDZ domains deleted does not. Loss of fas2 in follicle cells results in their abnormal rearrangement and invasion into the nurse cells [66, 67]. Together these results demonstrate that maintenance of properly organized apical and basolateral domains is essential to prevent the movement of cells out of an epithelium. Mutation of any number of genes that code for epithelial polarity proteins may therefore contribute to carcinoma metastasis.
By taking advantage of mutations in scrib and other tumor suppressor/polarity proteins, several labs have developed two different systems to generate marked tumors in vivo and then follow tumor cells that have moved to other locations (Fig. 2). In one example, tumor cells expressing a reporter gene such as beta-galactosidase are dissected from lgl or dlg mutant larvae and then transplanted into the abdomens of adult hosts (Fig. 2a). Later the marked cells can be identified even when they are far from the site of injection. Cells from mutant brains “metastasize” more frequently than cells taken from other larval tissues. The invasive cells are able to cross cell layers and basement membranes, and in some cases, proliferate. For example, some micrometastases were identified that had moved through a sheath composed of muscle cells and across basement membranes within the ovary [68]. Since metastatic tumor cells also cross multiple cell layers and basement membranes during their spread, the ability to model these steps in Drosophila may provide an opportunity to use genetics to identify additional genes required for these events.
In another series of transplantation experiments (Fig. 2a), Beaucher et al. [69] induced tumor formation using cells mutant for the translational repressor brain tumor (brat) and compared the metastatic behavior of lgl and brat mutant cells. While both lgl and brat mutant cells showed similar frequencies of metastatic spread when transplanted into a host female, they behaved differently following serial transplantations. Cells mutant for lgl that metastasized upon transplantation into a host female showed an increasing propensity to metastasize each time they were serially transferred to a new host, suggesting that the procedure was selecting for the most highly metastatic cells. In contrast, brat mutant cells exhibited a stable metastatic behavior upon serial transplantation, demonstrating that tumors caused by different initiating genetic alterations show different metastatic properties [69].
Woodhouse et al. [70], took advantage of the highly metastatic behavior of transplanted lgl mutant cells to carry out a genetic screen for modifiers of metastasis. They identified three genes that, when double mutant with lgl, modified the metastatic behavior of cells. Mutation of the semaphorin-5c gene dramatically reduced the growth and spread of lgl mutant cells following transplantation. Although this mutation was previously uncharacterized, the gene belongs to the semaphorin family, which is known for its function in axon guidance in both insects and mammals. SEMA-5c is a transmembrane protein with extracellular Thromospondin repeats. Two other genes that alter the behavior of lgl mutant cells were identified in this screen. An unusual allele of apontic that causes its overexpression resulted in a cell-autonomous loss of metastasis without affecting growth at the primary grafting site [70]. This protein has previously been shown to act as a transcription factor required during embryogenesis and as a translation factor during oogenesis [71, 72]. An activating mutation in the ETS-family transcription factor called pointed accelerated host lethality caused by lgl tumors. Pointed is required for embryonic development and is known to be downstream of the EGF receptor or Ras signaling.
A second approach that has been used to model metastatic disease is reported in two studies that took advantage of the many sophisticated genetic tricks in Drosophila to generate GFP-marked mutant clonal populations of cells expressing either a gain of function allele of Ras, RasV12, or homozygous for loss of function mutations in scrib while simultaneously altering additional genetic pathways (Fig. 2b). Brumby and Richardson made homozygous mutant clones of scrib initially, and found that these were outcompeted by neighboring cells and eventually died by Jun N-terminal kinase (JNK)-induced apoptosis—a situation that may occur in human tumorigenesis as well. In contrast, the authors showed that when Ras or Notch was activated, in addition to loss of scrib, the mutant clones survived and formed tumors. However, scrib mutant tissue that also had EGFR, Wingless (Wnt), or DPP (TGF-β) signaling blocked survived less well than scrib alone [73]. In a separate study, Pagliarini and Xu induced tumors by making clones overexpressing RasV12 and then screened for loss-of-function changes that altered the dissemination of labeled cells. They found that mutations in scrib, in the RasV12 background resulted in tumor cells that could invade neighboring cells. These tumors had lower DE-cadherin levels, and overexpression of DE-cadherin blocked their invasive behavior. Neighboring cells also showed alterations in collagen and laminin proteins in response to the metastatic cells, suggesting that the tumor cells could breakdown the extracellular matrix [74]. This type of “metastasis” is clearly different from that in mammals, as the adult fly does not have blood vessels, so cells do not spread by entering the bloodstream. However, many aspects of metastasis are conserved in these models, including dissociation from the primary tumor, invasion into other tissues, sometimes to distant sites, and changes in molecular markers of adhesion and ECM. More recently, it was discovered that activation of Raf can also lead to invasive behavior of scrib clones [75]. This work showed a non-autonomous effect of Raf and JNK on neighboring tissue. Another study recently demonstrated that JNK activation is probably a primary downstream mediator for tumor growth due to the loss of polarity proteins [76]; JNK pathway components are upregulated in these tumors, and a dominant negative form of JNK is sufficient to block growth. These types of studies clearly lend themselves to testing additional gene functions in the process of tumor metastasis.
Another strategy to understand metastasis is a reverse-genetic approach: to take genes identified as playing a role in metastasis in mammals and disrupt the functions of homologous genes in flies. Many mammalian gene families have only one or two Drosophila counterparts, so analysis of the signaling pathways is simplified. In addition, many genetic tools in flies are available to facilitate these analyzes. In the long-term, this strategy is likely to aid in unraveling complex genetic interactions and may clarify essential protein functions.
MMPs seem to play a major role in tumor metastasis, but with over 20 family members in mammals, functional redundancy poses a challenge in understanding their normal roles and interactions. The Drosophila genome encodes only two such proteins, and disruptions of both resulted in viable embryos but lethality later in development, partially attributed to disruptions in tracheal development, nervous system remodeling and epithelial fusion. MMP1 has been shown to associate with a cell adhesion molecule, Ninjurin A, which may help to explain its mechanism of action [77]. MMP1 causes NinjurinA to be cleaved, resulting in cell non-autonomous loss of cell adhesion due to the liberation of the NinjurinA extracellular N-terminal domain. In the clonal metastasis models described above using scrib loss-of-function to promote tumor formation, mmp1 was shown to be upregulated [78]. Furthermore, loss of mmp1 function by RNAi suppressed cell invasiveness [78]. As a corollary, the single fly tissue-inhibitor of matrix metalloproteases (TIMP-1), could also reduce invasiveness when overexpressed. Similar results were obtained from another lab, using the injected-tumor approach. Interestingly, mmp1 disruption reduces the metastatic potential of lgl −/− cells, but not that of brat mutant tissue [68]. In contrast, loss of mmp1 function in the host resulted in more ovarian metastases of transplanted tissue from either lgl or brat mutants. However, there was an increase in ovarian expression of mmp1 only in response to brat but not lgl tumors. These results demonstrate an important role for MMPs in metastasis in the fly and suggest that genetically distinct tumors alter their microenvironment in distinct ways.
In another recent study, investigators reported the effects of expressing mutated forms of human E-cadherin in cells of the developing Drosophila wing, to assess their effects in an in vivo model system [79]. As discussed earlier, E-cadherin is a critical cell–cell adhesion molecule in normal epithelia, and the gene coding for in E-cadherin is frequently mutated in a variety of human cancers. Many of these mutations are clear loss-of-function mutations. However sometimes there are point mutations for which it is not entirely clear if the effect is loss-of-function or gain-of-function, or even something else. Such is the case for mutations in the CDH1 gene that are associated with hereditary diffuse gastric cancer syndrome (HDGC). Germline mutations in the CDH1 gene have been shown to cause this disease and several point mutations have been identified. One mutation in the extracellular domain (A634V) and one in the intracellular domain (V832M) were shown to exert distinct effects on cultured CHO cells in cell migration and invasion assays in vitro. Pereira et al. [79] decided to investigate the effects of expressing these altered proteins in Drosophila epithelia. Expression of normal human E-cadherin did not disrupt the epithelial architecture of cells in the wing primordium, known as an imaginal disc. However expression of the altered forms caused cells to drop out of the epithelium and invade adjacent tissue. The two different mutations caused distinct cellular behaviors such that A634V cells moved in large connected groups whereas V832M cells escaped the epithelium in smaller groups and even as individual cells. These analyses were carried out on fixed tissue so there are limitations to interpreting precisely what the dynamic behavior of the cells was, and it would be interesting to monitor the behavior of these cells in living tissue to obtain more precise information regarding the effects of these altered E-cadherin molecules. In addition, these studies were carried out in cells expressing normal, Drosophila E-cadherin, in addition to the ectopically expressed human protein. To mimic the situation in human tumor cells better, it would be useful to observe the behavior of cells expressing only human isoforms of E-cadherin. This approach may turn out to be quite powerful because, again, Drosophila genetics can be applied to identify additional genes that either exacerbate or ameliorate the phenotypes.
Increasing evidence suggests that tumor development depends on not just one mutation, but many alterations in gene expression that act in concert to promote growth, survival, and motility. Molecular signatures, as identified by microarray analysis, are being characterized in motile fly cells as well as in metastatic cancers, and may reveal common patterns. However, the large numbers of novel and uncharacterized genes identified in this way demonstrates there is a tremendous amount of basic research still to be done. In light of this view, simplified in vivo model systems, in which proteins act during development as they might in tumors, become more valuable. Without question, the genetic tricks and screens available in Drosophila will continue to provide important insights into how pathways to metastasis come together, and will help to unravel the basic biology of both normal and pathological cell movements.
References
Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006;12 8:895–904.
Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature 1980;287 5785:795–801.
Evangelista M, Tian H, de Sauvage FJ. The hedgehog signaling pathway in cancer. Clin Cancer Res 2006;12 20 Pt 1:5924–8.
Leptin M, Grunewald B. Cell shape changes during gastrulation in Drosophila. Development 1990;110 1:73–84.
Yang J, Mani SA, Weinberg RA. Exploring a new twist on tumor metastasis. Cancer Res 2006;66 9:4549–52.
Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol 2003;15 6:740–6.
Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol 2005;17 5:548–58.
Gryzik T, Muller HA. FGF8-like1 and FGF8-like2 encode putative ligands of the FGF receptor Htl and are required for mesoderm migration in the Drosophila gastrula. Curr Biol 2004;14 8:659–67.
Stathopoulos A, Tam B, Ronshaugen M, Frasch M, Levine M. Pyramus and thisbe: FGF genes that pattern the mesoderm of Drosophila embryos. Genes Dev 2004;18 6:687–99.
Kunwar PS, Starz-Gaiano M, Bainton RJ, Heberlein U, Lehmann R. Tre1, a g protein-coupled receptor, directs transepithelial migration of Drosophila germ cells. PLoS Biol 2003;1 3:E80.
Renault AD, Sigal YJ, Morris AJ, Lehmann R. Soma-germ line competition for lipid phosphate uptake regulates germ cell migration and survival. Science 2004;305 5692:1963–6.
Metzger RJ, Krasnow MA. Genetic control of branching morphogenesis. Science 1999;284 5420:1635–9.
Kerman BE, Cheshire AM, Andrew DJ. From fate to function: the Drosophila trachea and salivary gland as models for tubulogenesis. Differentiation 2006;74 7:326–48.
Affolter M, Bellusci S, Itoh N, Shilo B, Thiery JP, Werb Z. Tube or not tube: remodeling epithelial tissues by branching morphogenesis. Dev Cell 2003;4 1:11–8.
Bruckner K, Kockel L, Duchek P, Luque CM, Rorth P, Perrimon N. The PDGF/VEGF receptor controls blood cell survival in Drosophila. Dev Cell 2004;7 1:73–84.
Cho NK, Keyes L, Johnson E, Heller J, Ryner L, Karim F, et al. Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell 2002;108 6:865–76.
Wood W, Faria C, Jacinto A. Distinct mechanisms regulate hemocyte chemotaxis during development and wound healing in Drosophila melanogaster. J Cell Biol 2006;173 3:405–16.
Stramer B, Wood W, Galko MJ, Redd MJ, Jacinto A, Parkhurst SM, et al. Live imaging of wound inflammation in Drosophila embryos reveals key roles for small GTPases during in vivo cell migration. J Cell Biol 2005;168 4:567–73.
Xia Y, Karin M. The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol 2004;14 2:94–101.
Vidal M, Cagan RL. Drosophila models for cancer research. Curr Opin Genet Dev 2006;16 1:10–6.
Naora H, Montell DJ. Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer 2005;5 5:355–66.
Silver DL, Geisbrecht ER, Montell DJ. Requirement for JAK/STAT signaling throughout border cell migration in Drosophila. Development 2005;132 15:3483–92.
Silver DL, Montell DJ. Paracrine signaling through the JAK/STAT pathway activates invasive behavior of ovarian epithelial cells in Drosophila. Cell 2001;107 7:831–41.
Montell DJ. Border-cell migration: the race is on. Nat Rev Mol Cell Biol 2003;4 1:13–24.
Montell DJ, Rorth P, Spradling AC. slow border cells, a locus required for a developmentally regulated cell migration during oogenesis, encodes Drosophila C/EBP. Cell 1992;71 1:51–62.
Wang X, Bo J, Bridges T, Dugan KD, Pan TC, Chodosh LA, et al. Analysis of cell migration using whole-genome expression profiling of migratory cells in the Drosophila ovary. Dev Cell 2006;10 4:483–95.
Liu Y, Montell DJ. Identification of mutations that cause cell migration defects in mosaic clones. Development 1999;126 9:1869–78.
McDonald JA, Pinheiro EM, Kadlec L, Schupbach T, Montell DJ. Multiple EGFR ligands participate in guiding migrating border cells. Dev Biol 2006;296:94–103.
McDonald JA, Pinheiro EM, Montell DJ. PVF1, a PDGF/VEGF homolog, is sufficient to guide border cells and interacts genetically with Taiman. Development 2003;130 15:3469–78.
Duchek P, Rorth P. Guidance of cell migration by EGF receptor signaling during Drosophila oogenesis. Science 2001;291 5501:131–3.
Ferretti G, Felici A, Papaldo P, Fabi A, Cognetti F. HER2/neu role in breast cancer: from a prognostic foe to a predictive friend. Curr Opin Obstet Gynecol 2007;19 1:56–62.
Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res 2005;65 12:5278–83.
Xue C, Liang F, Mahmood R, Vuolo M, Wyckoff J, Qian H, et al. ErbB3-dependent motility and intravasation in breast cancer metastasis. Cancer Res 2006;66 3:1418–26.
de Launoit Y, Baert JL, Chotteau-Lelievre A, Monte D, Coutte L, Mauen S, et al. The Ets transcription factors of the PEA3 group: transcriptional regulators in metastasis. Biochim Biophys Acta 2006;1766 1:79–87.
Schober M, Rebay I, Perrimon N. Function of the ETS transcription factor Yan in border cell migration. Development 2005;132 15:3493–504.
Niewiadomska P, Godt D, Tepass U. DE-Cadherin is required for intercellular motility during Drosophila oogenesis. J Cell Biol 1999;144 3:533–47.
Bai J, Uehara Y, Montell DJ. Regulation of invasive cell behavior by taiman, a Drosophila protein related to AIB1, a steroid receptor coactivator amplified in breast cancer. Cell 2000;103 7:1047–58.
Pinheiro EM, Montell DJ. Requirement for Par-6 and Bazooka in Drosophila border cell migration. Development 2004;131 21:5243–51.
Montell DJ. Command and control: regulatory pathways controlling invasive behavior of the border cells. Mech Dev 2001;105 1–2:19–25.
Vleminckx K, Vakaet L, Jr., Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991;66 1:107–19.
Berx G, Cleton-Jansen AM, Strumane K, de Leeuw WJ, Nollet F, van Roy F, et al. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 1996;13 9:1919–25.
Cowin P, Rowlands TM, Hatsell SJ. Cadherins and catenins in breast cancer. Curr Opin Cell Biol 2005;17 5:499–508.
Geisbrecht ER, Montell DJ. Myosin VI is required for E-cadherin-mediated border cell migration. Nat Cell Biol 2002;4 8:616–20.
Yoshida H, Cheng W, Hung J, Montell D, Geisbrecht E, Rosen D, et al. Lessons from border cell migration in the Drosophila ovary: A role for myosin VI in dissemination of human ovarian cancer. Proc Natl Acad Sci U S A 2004;101 21:8144–9.
Dunn TA, Chen S, Faith DA, Hicks JL, Platz EA, Chen Y, et al. A novel role of myosin VI in human prostate cancer. Am J Pathol 2006;169 5:1843–54.
Carney GE, Bender M. The Drosophila ecdysone receptor (EcR) gene is required maternally for normal oogenesis. Genetics 2000;154 3:1203–11.
Cherbas L, Hu X, Zhimulev I, Belyaeva E, Cherbas P. EcR isoforms in Drosophila: testing tissue-specific requirements by targeted blockade and rescue. Development 2003;130 2:271–84.
Torres-Arzayus MI, Yuan J, DellaGatta JL, Lane H, Kung AL, Brown M. Targeting the AIB1 oncogene through mammalian target of rapamycin inhibition in the mammary gland. Cancer Res 2006;66 23:11381–8.
Kuang SQ, Liao L, Wang S, Medina D, O’Malley BW, Xu J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res 2005;65 17:7993–8002.
Yoshida H, Liu J, Samuel S, Cheng W, Rosen D, Naora H. Steroid receptor coactivator-3, a homolog of Taiman that controls cell migration in the Drosophila ovary, regulates migration of human ovarian cancer cells. Mol Cell Endocrinol 2005;245 1–2:77–85.
Oh A, List HJ, Reiter R, Mani A, Zhang Y, Gehan E, et al. The nuclear receptor coactivator AIB1 mediates insulin-like growth factor I-induced phenotypic changes in human breast cancer cells. Cancer Res 2004;64 22:8299–308.
Roy M, Pear WS, Aster JC. The multifaceted role of Notch in cancer. Curr Opin Genet Dev 2007;17 1:52–9.
Edenfeld G, Volohonsky G, Krukkert K, Naffin E, Lammel U, Grimm A, et al. The splicing factor crooked neck associates with the RNA-binding protein HOW to control glial cell maturation in Drosophila. Neuron 2006;52 6:969–80.
Lopez-Schier H, St Johnston D. Delta signaling from the germ line controls the proliferation and differentiation of the somatic follicle cells during Drosophila oogenesis. Genes Dev 2001;15 11:1393–405.
Wang X, Adam JC, Montell D. Spatially localized Kuzbanian required for specific activation of Notch during border cell migration. Dev Biol 2007;301 2:532–40.
Callahan R, Egan SE. Notch signaling in mammary development and oncogenesis. J Mammary Gland Biol Neoplasia 2004;9 2:145–63.
Humbert PO, Dow LE, Russell SM. The Scribble and Par complexes in polarity and migration: friends or foes? Trends Cell Biol 2006;16 12:622–30.
Dow LE, Kauffman JS, Caddy J, Peterson AS, Jane SM, Russell SM, et al. The tumour-suppressor Scribble dictates cell polarity during directed epithelial migration: regulation of Rho GTPase recruitment to the leading edge. Oncogene 2007;26:2272–82.
Grifoni D, Garoia F, Schimanski CC, Schmitz G, Laurenti E, Galle PR, et al. The human protein Hugl-1 substitutes for Drosophila lethal giant larvae tumour suppressor function in vivo. Oncogene 2004;23 53:8688–94.
Handa K, Yugawa T, Narisawa-Saito M, Ohno S, Fujita M, Kiyono T. E6AP-Dependent degradation of DLG4/PSD95 by high-risk human papillomavirus type 18 E6 protein. J Virol 2007;81 3:1379–89.
Bilder D. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev 2004;18 16:1909–25.
Nagasaka K, Nakagawa S, Yano T, Takizawa S, Matsumoto Y, Tsuruga T, et al. Human homolog of Drosophila tumor suppressor Scribble negatively regulates cell-cycle progression from G1 to S phase by localizing at the basolateral membrane in epithelial cells. Cancer Sci 2006;97 11:1217–25.
Nguyen MM, Nguyen ML, Caruana G, Bernstein A, Lambert PF, Griep AE. Requirement of PDZ-containing proteins for cell cycle regulation and differentiation in the mouse lens epithelium. Mol Cell Biol 2003;23 24:8970–81.
Goode S, Perrimon N. Brainiac and fringe are similar pioneer proteins that impart specificity to Notch signaling during Drosophila development. Cold Spring Harb Symp Quant Biol 1997;62:177–84.
Abdelilah-Seyfried S, Cox DN, Jan YN. Bazooka is a permissive factor for the invasive behavior of discs large tumor cells in Drosophila ovarian follicular epithelia. Development 2003;130 9:1927–35.
Szafranski P, Goode S. A Fasciclin 2 morphogenetic switch organizes epithelial cell cluster polarity and motility. Development 2004;131 9:2023–36.
Szafranski P, Goode S. Basolateral junctions are sufficient to suppress epithelial invasion during Drosophila oogenesis. Dev Dyn 2007;236 2:364–73.
Beaucher M, Hersperger E, Page-McCaw A, Shearn A. Metastatic ability of Drosophila tumors depends on MMP activity. Dev Biol 2007;303 2:625–34.
Beaucher M, Goodliffe J, Hersperger E, Trunova S, Frydman H, Shearn A. Drosophila brain tumor metastases express both neuronal and glial cell type markers. Dev Biol 2007;301 1:287–97.
Woodhouse EC, Fisher A, Bandle RW, Bryant-Greenwood B, Charboneau L, Petricoin EF, 3rd, et al. Drosophila screening model for metastasis: semaphorin 5c is required for l(2)gl cancer phenotype. Proc Natl Acad Sci U S A 2003;100 20:11463–8.
Eulenberg KG, Schuh R. The tracheae defective gene encodes a bZIP protein that controls tracheal cell movement during Drosophila embryogenesis. EMBO J 1997;16 23:7156–65.
Lie YS, Macdonald PM. Apontic binds the translational repressor Bruno and is implicated in regulation of oskar mRNA translation. Development 1999;126 6:1129–38.
Brumby AM, Richardson HE. Using Drosophila melanogaster to map human cancer pathways. Nat Rev Cancer 2005;5 8:626–39.
Pagliarini RA, Xu T. A genetic screen in Drosophila for metastatic behavior. Science 2003;302 5648:1227–31.
Uhlirova M, Jasper H, Bohmann D. Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc Natl Acad Sci U S A 2005;102 37:13123–8.
Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol 2006;16 11:1139–46.
Zhang S, Dailey GM, Kwan E, Glasheen BM, Sroga GE, Page-McCaw A. An MMP liberates the Ninjurin A ectodomain to signal a loss of cell adhesion. Genes Dev 2006;20 14:1899–910.
Uhlirova M, Bohmann D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J 2006;25 22:5294–304.
Pereira PS, Teixeira A, Pinho S, Ferreira P, Fernandes J, Oliveira C, et al. E-cadherin missense mutations, associated with hereditary diffuse gastric cancer (HDGC) syndrome, display distinct invasive behaviors and genetic interactions with the Wnt and Notch pathways in Drosophila epithelia. Hum Mol Genet 2006;15 10:1704–12.
Michelson AM, Gisselbrecht S, Zhou Y, Baek KH, Buff EM. Dual functions of the heartless fibroblast growth factor receptor in development of the Drosophila embryonic mesoderm. Dev Genet 1998;22 3:212–29.
Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell 2001;107 1:17–26.
Acknowledgment
A. C. C. Jang, M. Starz-Gaiano, and D. J. Montell contributed equally to this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jang, A.CC., Starz-Gaiano, M. & Montell, D.J. Modeling Migration and Metastasis in Drosophila . J Mammary Gland Biol Neoplasia 12, 103–114 (2007). https://doi.org/10.1007/s10911-007-9042-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-007-9042-8