
Abstract
In this paper, we investigate the ion rejection of salt water using graphene sheet as a semi-permeable membrane. Both the mathematical modeling and MD simulations will be performed to determine the acceptance conditions for a water molecule or a sodium ion permeating into the membrane. Chloride ion is always blocked by the graphene due to the fact that the ionic size of the chloride ion is larger than the pore size of the graphene leaving the sieve of water and sodium ions which depends on the strength of the external forces. In particular, certain ranges of the external forces will be theoretically deduced for the complete desalination, which turn out to depend intimately on the size of the permeate container and the hydraulic force acting among salt water. In this paper, we reduce the multi-body system into several two-body systems and reduce the 3D problem into degenerated 1D problems using the continuous approximation, where the molecular interactions between the water molecule or the sodium ion and the graphene could be determined in terms of surface integrals. Given the force fields between the intruder and the membrane, MD simulations could be used to investigate the time evolution of the system and compare with the theoretical results deduced by the present mathematical model. We confirm the computational results given by Tanugi and Grossman (Nano Lett 12:3602–3608, 2012). Moreover, our approach is computationally rapid and generates inductive results for more engineering applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Numerous computational simulations on ultra-filtration using nanotubes [2–9] and zeolite [10] have been reported in current literature. All of these studies reveal the rapid and efficient ultra-filtration using nanomaterials embedded inside a polymer matrix in comparison to conventional membranes. Nanomaterial related-membranes can be used as semi-permeable membranes which allow certain molecules or ions to pass through but expel heavier molecules or ions by the mean of reverse osmosis, herein RO. For example, upon compressing seawater by an applied force towards a carbon nanotube membrane, the membrane could accept water but repel both the sodium and chloride ions. Moreover, an interior of small-radii carbon nanotubes provides the hydrophobic surface so that a rapid conduction of water molecules inside nanotubes as the form of single-file transport is feasible [6, 7, 11–13]. RO has found to possess merits of low energy consumption and operational costs in comparison to other desalination methods such as multi-stage flash, multi-effect distillation and mechanical vapor compression. Although numerous theoretical investigations of using such nanomaterials for the ultra-filtration have been performed, none of these nanomaterials have successful been proved for the desalination. For instance, it is very hard to produce massive and highly aligned carbon nanotube arrays although few methods have been recently reported using thermocapillary flows [14] and the water permeability inside zeolite is low which hampers the conduction of water inside zeolite [1]. Recently, Tanugi and Grossman [1] have proposed using nano-porous graphene for the seawater desalination due to the high mechanical strength and the negligible thickness of the graphene sheet, which is a 2D sheet consisting of \(sp^{2}\) bonded carbon atoms arranged in the form of hexagonal honeycomb lattice (See Fig. 1 for details). They also claim that graphene possesses the highest water permeability in comparison to the existing commercial RO and MFI zeolite [1]. Graphene has unique mechanical, electronic, magnetic and optical properties which potentially opens up numerous applications for future devices, and we refer our readers to [15] for some recent advances in graphene. In addition, several experimental investigations have shown that the possibility of producing and transferring a single layered graphene sheet [16], which makes this 2D-sheet more applicable for the desalination.

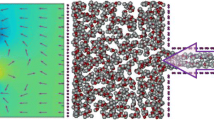
A computational simulation of desalination using nano-porous graphene, where the left-hand side is pure water while the right-hand side is salt water (Copyright @MIT)
However, the simulation approach adopted by Tanugi and Grossman [1] and other similar computational simulations [2–10] have severe temporal and spatial constraints that they are infeasible to take into account these multi-body problems in long run and large size. Here, we adopt the continuous approximation introduced by Cox et al. [17, 18] to coarse grain the pairwise interactions between molecules and ions in order to reduce the multi-body system into several two-body systems and reduce the 3D problem into the corresponding degenerated 1D problems so that the time-consuming pairwise calculations could be approximated by solving single or double surface integrals, i.e. Eqs. (2) and (3). Continuous approximation has widely been used for many applications at the nanoscale including the buckling analysis of carbon nanotubes [19, 20], biological applications including drug and gene deliveries [21–23], energy storage inside graphene [24, 25], gas storage inside nanomaterials [26–28] and computational memory devices [29]. More recently, the present author has extended the methodology to investigate the single-file transport of water molecules inside carbon nanotubes [11], massive hydrogen yield using functionalized nanomaterials [30], hydrogen storage inside doubly-layered graphene [31] and the ultra-filtration using functionalized carbon nanotubes [12, 13, 32]. Given the force fields between molecules and ions, the Verlet algorithm [33] could also be used to determine the time evolution of such mechanical systems.
In this paper, we explore the possibility and limitation of using the graphene sheet for desalination purposes. We assume that the carbon atoms are smeared across the surface of the graphene sheet, and the oxygen, the sodium ion and the chloride ion are smeared across an envisaged surface of radius 1, 1.16 and 1.67 Å, respectively so that the molecular interactions between any two rigid bodies could be approximated by surface integrals, which could be incorporated into the Verlet algorithm so that the time evolution of the mechanical system could be determined numerically. In Sect. 2, we lay out the theoretical and computational basis for the rest of the paper and the corresponding numerical results are presented in Sect. 3. A general conclusion is provided in the final section of the paper Table 1.
2 Theory and MD simulations
In this section, we introduce the theoretical and algorithmic basis for the rest of the paper. For simplicity, we model the molecular interactions between a water molecule or a ion and the graphene by the usual 6–12 Lennard–Jones potential [34]
where \(\rho \), \(\epsilon \), \(\sigma \), A and B denote the atomic distance between two typical atoms, the Lennard–Jones potential well depth, the Lennard–Jones distance between two atoms, the attractive constant and the repulsive constant, respectively. In addition, the steric effects could be captured by the repulsive term, i.e. \(1/\rho ^{12}\) term appearing in Eq. (1). To generate accurate results, more intricate empirical potentials such as the morse potential and TIP4P potential could be used but this limits the feasibility of partially solving the surface integrals, i.e. Eqs. (2) and (3) analytically resulting in longer computational times. The numerical values of several constants including the pore size of graphene sheet are summarized in Table 2. We assume that the carbon atoms are smeared across the graphene sheet where we approximate the hexagonal pores by circles of radii 1.42 Å only for the sake of simpler mathematical derivation, and the oxygen atom and the sodium and chloride ions are smeared across on an envisaged sphere of radius 1, 1.16 and 1.67 Å, respectively so that the van der Waals forces between the oxygen or the sodium and chloride ions and the graphene could be approximated by the double surface integral [17, 18] which is given by
where \(\eta _{1}\), \(\eta _{2}\), \(-dV(\rho )/d\rho \mid _{\perp }\), \(dS_{1}\) and \(dS_{2}\) denote the number density of the graphene sheet, the number density of the envisaged sphere, the axial force generated by the potential energy given in Eq. (1), the surface element of the graphene sheet and the surface element of the envisaged sphere, respectively. We comment that the subscript 2 of \(F_{2}\) indicates the double surface integral and the aftermath of series of calculations ends up in a function of R only, which is an axial distance between the center of the intruder and the pore center (See Fig. 2). In addition, the interactions between the hydrogen and the graphene is determined by the single surface integral which is given by

Coordinates of a water molecule intruding into a nearest pore (solid hexagon on the graphene sheet), where \(\rho \) denotes the atomic distance between a hydrogen atom and an arbitrary point on the graphene sheet. Both the applied and hydraulic forces are shown in terms of different arrows. We note that due to the compactness and the homogeneity of pores on the graphene sheet, it is legitimate to say that the average axial force fields generated on the water molecule are roughly the same on the envisaged plane which is parallel with the graphene
Both the single and double surface integrals reduce the multi-body interactions into several two-body interactions and the introduction of the axial forces reduce the 3D problem into degenerated 1D problems. In principle, such integrations could be solved numerically using Monte–Carlo method which incurs huge computational times however according to the prior works done by the present author [12, 13, 32], certain parts of the surface integrals could be solved analytically facilitating rapid computational times. The molecular interactions between the water molecule and the graphene is therefore the linear sum of one double surface integral, i.e. oxygen and graphene, and two single surface integrals, i.e. two hydrogens and graphene. Since the orientation of water molecules is found to be crucial in this problem [35], we initially fix the configuration of the two hydrogen atoms separated by an angle \(2\alpha \) as shown in Fig. 2 and different orientations of the water molecule are generated by two independent angles \(\theta \in [0,\pi ]\) and \(\psi \in [-\pi ,\pi ]\), so that the distance between the first hydrogen (Upper hydrogen in Fig. 2) and an arbitrary point on the graphene, \(\rho \) is written as
where a, \(\ell \), R, \(\phi \) and \(\Phi \) denote the radius of the water molecule, the distance between the center of the pore and an arbitrary point on the graphene sheet, the distance between the centers of the water molecule and the pore center, half of the angle between two hydrogens and the polar angle on the graphene, respectively (See Fig. 2 for details). Due to the symmetry of two hydrogen atoms, the distance between the second hydrogen and the graphene can be obtained by replacing \(\phi \) with \(-\phi \). The distance between the oxygen or the sodium and chloride ions and the graphene sheet can also be deduced in a similar manner. Such atomic distances must be incorporated into Eqs. (2) and (3) to determine \(F_{1}\) and \(F_{2}\). The total force between the water molecule and the graphene is therefore written as
where \(F_{O}\) denotes the force between the oxygen and the graphene and its calculation is given by Eq. (2), \(F_{H_{1}}\) and \(F_{H_{2}}\) denote the force between the first hydrogen and the second hydrogen and the graphene, respectively and their calculations are given by Eq. (3), \(F_{hydra}\) denotes the hydraulic forces in the solute, \(F_{app}\) denotes the applied force, \(F_{vdW}\) denotes the van der Waals forces between the water molecule and the graphene, and \(F_{ext}\) denotes the external forces which consist of the hydraulic and applied forces acting perpendicularly to the graphene sheet. This equation describes the average force fields experienced by a single water molecule. A particular attention should be paid here that even we are practically considering a normal graphene with many pores (see Fig. 1 for details). However, due to the compactness of the pores on the graphene sheet and the homogeneity of the graphene system, there exists the translational invariance of the water molecule or the sodium ion on the envisaged planes that are parallel with the graphene sheet, where the average axial force fields are roughly the same on the planes (See Fig. 2 for details). Therefore, we can reduce the current 3D system into degenerated two-body systems, where the single degenerated “pore” investigated here represents the nearest pore encountered by a single water molecule which could be changed temporally when the it approaches the graphene. In addition, near the pore proximity, the trajectory turns out to be a straight line due to the confinement of molecular forces arising from the pore entry. We also comment that we coarse grain the external forces \(F_{ext}\) by the lump sum of both the hydraulic and applied forces and the value of hydraulic forces could be obtained by either experimental data or simulation results. In this paper, we will attempt different forms of \(F_{ext}\) to gain some physical insights for the ultra-filtration problem.
Since \(\left\{ F_{H_{i}}|i=1,2\right\} \) are generated by different orientations, the ensemble forces acting on the two hydrogens could be determined using the Boltzmann’s distribution
where j, \(F_{H_{i}}(R)_{j}\), \(\beta \) and \(V_{H_{i}}(R)_{j}\) denote the different orientations, forces of \(H_{i}\) in j configuration calculated using Eq. (3), the reciprocal of the usual Boltzmann’s constant times the temperature, and the potential energy between \(H_{i}\) and the graphene in j configuration which can be obtained using Eq. (3) by replacing \(-dV(\rho )/d\rho \mid _{\perp }\) with \(V(\rho )\), respectively. Temperature effect is thus absorbed in Eq. (6). We will observe later that the internal energy will be raised during the compression process. The total force between the ions and the graphene sheet can be deduced in a similar fashion, i.e. \(F^{tot}=F_{I}+F_{ext}\), where \(F_{I}\) denotes the van der Waals forces between the ions and the graphene, which is calculated using Eq. (2) and \(F_{ext}=F_{hydra}+F_{app}\). Given these force fields, the time evolution of the system can be numerically determined using Verlet algorithm
where \(\tau \), k, M and V denotes the time grid, the time step, the mass of the intruder and the velocity fields, respectively. To facilitate more accurate numerical outcomes, multi-step technique [36] will be used in the significant force regime.
3 Numerical results and discussion
In this section, we carry out some numerical results as given in Section 2. Two subsections will be added to demonstrate the inductive nature of the present methodology. Inserting the value of parameters given in Table 2 into Eqs. (2–6) and assuming \(T=300\) K, we determine the Van der Waals forces of the water molecule with 100 different orientations and the sodium ion permeating into the graphene, which are plot together in Fig. 3 for comparison.

Van der Waals forces of the water molecule and the sodium ion entering into the graphene where the positive forces, the vertical block line at \(R=0\) Å and the red sphere imply the repulsive forces generated by the graphene, the location of the graphene and the intruder, respectively. The Unit of R is Å
Due to the fact that the molecular size of the chloride ion is larger than the pore size of the graphene, the chloride ion always repels from the sheet by Pauli exclusive principle. From Fig. 3, both the water molecule and the sodium ion experience repulsive forces but occurring in different magnitudes when they approach the graphene sheet. Without any external forces, both the water molecule and the sodium ion are blocked by the graphene. While the external forces are applied into the system, the salt water will be pushed towards the sheet in order to overcome the repulsive forces generated by the graphene. Since the repulsive forces of the sodium ion are stronger than that of the water molecule, there must exist a range of external forces, so that for a given container size, the water molecule will get through the graphene sheet while the sodium ion will be rejected from the graphene. Once the water molecule or the sodium ion reaches the center of the graphene, the repulsive forces generated from the other side of the graphene will automatically suck them out of the sheet so that the acceptance criteria for the desalination could be written as
where \(R_{0}\), \(F_{vdW}^{I}\) and \(F_{vdW}^{W}\) denote the farthest possible initial position of the intruder, the van der Waals forces for the sodium ion and the van der Waals forces for the water molecule, respectively. Here, we might assume \(R_{0}\) to be the size of the permeate container as we purposely consider the axial forces in Eqs. (2) and (3). In addition, if an intruder could not pass through the graphene from the container end by the external forces, it is certainly not able to do so everywhere inside the container. Such inequality is valid if and only if the integral from the left hand side is larger than that from the right hand side. For the constant \(F_{ext}\), Eq. (8) reduces to
We could interpolate data from Fig. 3 and calculate the integrations in Eq. (9) using trapezoidal rule. We comment that for a given \(R_{0}\), Eq. (9) provides predicted values for the range of the external forces, for which the 100 % seawater desalination is possible and such ranges are given in the middle column of Table 1.
For constant \(F_{ext}\), we observe that the larger the container size, the smaller the external forces are required to squeeze water or sodium ions through the graphene as more work has been done on them during RO. Since the molecular forces between the water molecule or the sodium ion and the graphene sheet diminish over distance, for sufficiently large \(R_{0}\), the range of the external forces depends reversely on \(R_{0}\). For example, the the range for \(R_{0}=20\) could be obtained by multiplying the range for \(R_{0}=10\) by 10 and then divided by 20. Moreover, the width of the ranges shrinks when the size of the container increases which are given in Fig. 4.

The width of the ranges for the proposed \(R_{0}\)
We observe from Fig. 4 that the width of the ranges drops exponentially for small \(R_{0}\) and becomes linear for \(R_{0}>20\) which poses some difficulties of using the graphene sheet for the commercial RO, especially when thermal fluctuation is taken into account. When the size of the container becomes so large that the complete desalination is almost impossible unless additional repulsive forces are generated at the pore entry, for instance by chemically attaching certain functional groups on the graphene sheet, termed graphene oxide frameworks. Upon assuming the homogeneity of the solute and the usual concentration of sodium ions in seawater, under the constant \(F_{ext}\), i.e. \(7 \times 10^{-4}\) eV/Å is applied to the container of size \(1\times 10^{3}\) Å\(^{3}\), which is equivalent to \(R_{0}=10\) Å. Such external forces are situated outside the corresponding range given in Table 1 and our theoretical calculations show that 97.2 % desalination rate could be reached. Without loss of generality, different forms of external forces could be taken and the acceptance external forces can be determined in a similar way for the desalination. Now we fix the size of the container by 10\(\times \)10 \(\times \) 10 Å\(^{3}\) which is about the spatial dimension of the usual computational simulations and we assume that the thickness of graphene sheet is negligible. From Fig. 3, we also assign the cut off distances for the molecular forces as \(-0.5\) and 1 Å to reduce our computational times. In addition, due to the significant molecular interactions occurring in \(R\in [-0.5, 1]\), time grid in this regime will be tuned 10 times finer than that in the bulk solute. Now, we let \(R_{0}=10\) Å and zero initial velocity (water molecules in the initial position undergo Brownian motion under thermal equilibrium resulting in zero net velocity), and assume \(F_{ext}\) vanishes on the other side of the graphene, we then incorporate the force fields, \(F_{tot}\) into the Verlet algorithm as stated in Eq. (7). The numerical results for five different constant external forces, i.e. 3, 4, 5, 6 and 7 \(\times \,10^{-4}\) eV/Å are shown in Figs. 5 and 7 for the water molecule and the sodium ion, respectively.

MD simulations for the water molecule permeating into graphene from 10 Å for five different external forces

Velocity field for \(R_{0}=10\) Å and \(F_{ext}=4 \times 10^{-4}\) eV/Å
Figures 5 and 7 might initially appear odd that unlike the usual MD simulations which provide the full dynamical picture of the entire system; here the present MD results only serve as an indicator, from which given the initial position, the numerical results only provide the time evolution of the axial motion of the water molecule and the sodium ion to its nearest pores which could be changed when the intruder approaches the graphene. From Fig. 5, we observe that except \(F_{ext}=3\times 10^{-4}\) eV/Å, the water molecule passes through the graphene for other proposed external forces. There is a kink occurring near the pore vicinity indicating a sudden push-out force and then a pull-in force for the accepted water molecule, which is induced by the repulsive forces given in Fig. 3. For \(R_{0}=10\) Å and \(F_{ext}=4\times 10^{-4}\) eV/Å, the velocity of the permeating water molecule given in Fig. 6 assures such phenomenon, where the velocity of the water molecule is slowed down when it approaches the graphene entry and it regains the strength of its velocity after passing through the graphene. An increase in the water velocity indicates the rise in the internal energy and the temperature of the system during RO. This enhances the reliability of the desalination using the graphene as a membrane. We also observe from Fig. 5 that the larger the external forces, the quicker the flow time is, where an unit time is equivalent to 10 fs.

MD simulations for the sodium ion permeating into graphene from 10 Å for five different external forces
On contrary, from Fig. 7, the sodium ion repels from the pore for \(F_{ext}=3, 4\) and \(5\times 10^{-4}\) eV/Å and is accepted into the graphene when \(F_{ext}\) is bigger than 6 \(\times 10^{-4}\) eV/Å so that for \(F_{ext}=4\) and \(5\times 10^{-4}\) eV/Å, only water can get through the graphene sheet but sodium ions are rejected by the sheet, which is consistent with the theoretical results predicted in the middle column of Table 1. The only discrepancy between the theoretical results and the simulations occurs at \(F_{ext}=6\) eV/Å, which could be partially explained by the combination of the adoption of the cut-off forces, and the round-off errors occurring in evaluating numerical integrations and performing the MD simulations. We also observe a kink for the accepted sodium ion which could be explained by the similar reasons as given for the case of the water molecule.
We comment that the innovation of the present methodology is that the computational times are dramatically reduced and the spatial constraint is largely eradicated. In usual MD simulations for such complex multi-body system, when a parameter, for example the applied pressure, the system size and the temperature is altered, the numerical results have to be obtained from the very beginning resulting in considerably long times. This limits the inductive power of usual MD simulations and the present methodology provides more engineering approaches towards commercial applications. We are in no way to dispute the more powerful and detailed outcomes offered by the usual MD simulations and the present methodology could form an alternative approach to yield further physical insights. Here, two scenarios, namely the desalination of different container sizes for the same external forces and the desalination using exponentially decayed external forces will be performed to demonstrate the inductive power of the present methodology in the following subsections.
3.1 Scenario one
Now, we use the same parameters given in above MD simulations and fix the external forces to 5 \(\times 10^{-4}\) eV/Å but we vary the size of the container. The numerical results of the water molecule and the sodium ion permeating into the graphene for five different initial positions, namely \(R_{0}=5\), 8, 10, 20 and 30 Å are given in Figs. 8 and 9, respectively.

MD simulations for the water molecule permeating into the graphene with \(F_{ext}=5\times 10^{-4}\) eV/Å for five proposed initial positions

MD simulations for the sodium ion permeating into the graphene with \(F_{ext}=5\times 10^{-4}\) eV/Å for five proposed initial positions
We observe from Figs. 8 and 9 that for the given external force, i.e. \(F_{ext}=5\times 10^{-4}\) eV/Å, the larger the initial position or the container size, the easier for the water molecule and the sodium ion to get through the graphene sheet. Note that the numerical results should be forfeited when R is larger than \(R_{0}\) and such numerical results are entirely consistent with the theoretical results presented in Table 1. In this case study, we obtained all numerical results in less than half hour and once we know the external forces, we could determine the range of container size, for example between 8 and 10 Å in this case, for the complete desalination.
3.2 Scenario Two
Often solutes are compressible and we should better consider an exponentially decayed external forces for example: \(F_{ext}=F_{hydra}+F_{app}\left\{ 1-\exp (-\beta R)\right\} \), where \(F_{hydra}\), \(F_{app}\) and \(\beta >0\) denote the hydraulic force, the applied force and a decay constant, respectively. Under this form, \(F_{app}\) diminishes exponentially towards the pore vicinity. \(F_{hydra}\) could be obtained using experimental data or MD simulations. Here, we simply assume a constant \(F_{hydra}=5\times 10^{-4}\) eV/Å, which is interpolated from Corry [4]. We comment that the value and form assumed for \(F_{hydra}\) might not be exact for our present study but at least at the right magnitude in comparison to the true value. We aim to investigate more accurate form for such force either analytically or computationally in our future study. Without the loss of generality, we let the decay constant be one and the acceptance applied force is given by

MD simulations for the water molecule and the sodium ion permeating into graphene with \(F_{app}=0.5\times 10^{-4}\) eV/Å and \(F_{hydra}=5\times 10^{-4}\) eV/Å
The numerical results are summarized in the last column of Table 1. We comment that the negative values of \(F_{app}\) imply the spontaneous suck in of the water molecule or the sodium ion without any applied force and the spontaneous suck in of the water molecule or the sodium ion depends on the side of Eq. (10) from which these negative values occur. For instance, for \(R_{0}=8\) Å, the negative value occurs at the left-hand side of the inequality which implies the spontaneous suck in of the water molecule whereas the sodium ion will pass through the graphene not until the applied force is bigger than 2.923 eV/Å. For \(R_{0}=30\) Å, both the water molecule and the sodium ion could get through the graphene without any applied force. We note that the width of all ranges is smaller than that of constant external forces due to the existence of the hydraulic forces. According to Table 1, spontaneous suck in of both the water molecule and the sodium ion occurs for \(R_{0}>20\) Å which implies that given \(F_{hydra}=5 \times 10^{-4}\) eV/Å, complete desalination is impossible when \(R_{0}>20\) Å. For example, without any applied force, for \(R_{0}=20\) Å, the maximum desalination rate could only reach 92 %. For \(R_{0}=10\), we observe that an applied force of 0.5 \(\times 10^{-4}\) eV/Å will accept water but repel sodium ions, which is assured by the MD simulations given in Fig. 10.
4 Conclusion
In this paper, we adopt both the continuous approximation and MD simulations to investigate the ultra-filtration of seawater using the graphene sheet, which reduce the 3D multi-body problem into certain degenerated 1D two-body problems. We find that for certain ranges of the external forces, the graphene accepts only water but repels sodium ions resulting in the seawater desalination. Both cons and pos of using graphene sheets for the desalination have been discussed, and the present methodology has the merit of rapid computational spread and generates inductive results. Simple Lennard–Jones potential is adopted in this paper due to the fact that certain integrations could be determined analytically facilitating rapid computational times. In principle, Lennard–Jones potential could be easily replaced by other more sophisticated empirical potential energies resulting in longer computational times. Least but not last, the validity of the paper is subject to future experimental and computational outcomes.
References
D.C. Tanugi, J.C. Grossman, Water desalination across nanoporous graphene. Nano Lett. 12, 3602–3608 (2012)
T. Hilder, D. Gordon, S. Chung, Salt rejection and water transport through boron nitride nanotubes. Small 5, 2183–2190 (2009)
G. Hummer, J.C. Rasaiah, J.P. Noworyta, Water conduction through the hydrophobic channel of a carbon nanotube. Nature 414, 188–190 (2001)
B. Corry, Designing carbon nanotube membranes for efficient water desalination. J. Phys. Chem. B 112, 1427–1434 (2008)
C. Song, B. Corry, Intrinsic ion selectivity of narrow hydrophobic pores. J. Phys. Chem. B 113, 7642–7649 (2009)
A. Berezhkovskii, G. Hummer, Single-file transport of water molecules through a carbon nanotube. Phys. Rev. Lett. 89, 064503 (2002)
G. Zuo, R. Shen, S. Ma, W. Guo, Transport properties of single-file water molecules inside a carbon nanotube biomimicking water channel. ACS Nano 4, 205–210 (2010)
A. Kalra, S. Garde, G. Hummer, Osmotic water transport through carbon nanotube membranes. PNAS 100, 10175–10180 (2003)
M.S.P. Sansom, I.H. Shrivastava, K.M. Ranatunga, G.R. Smith, Simulations of ion channels watching ions and water move. Trends Biochem. Sci. 25, 368–374 (2000)
M. Theresa, M. Pendergast, E.M.V. Hoek, A review of water treatment membrane nanotechnologies. Energy Environ. Sci. 4, 1946–1971 (2011)
Y. Chan, J.M. Hill, A mechanical model for single-file transport of water through carbon nanotube membranes. J. Membr. Sci. 372, 57–65 (2011)
Y. Chan, J.M. Hill, Modeling on ion rejection using membranes comprising ultra-small radii carbon nanotubes. Eur. Phys. J. B 85, 56 (2012)
Y. Chan, Mathematical modeling on ultra-filtration using functionalized carbon nanotubes. Appl. Mech. Mater. 51, 1258–1273 (2013)
H.J. Sung et al., Using nanoscale theromocapillary flows to create arrays of purely semiconducting single-walled carbon nanotubes. Nat. Nanotechnol. 8, 347–355 (2013)
W.A.D. Heer, C. Berger, Editorial: epitaxial graphene. J. Phys. D Appl. Phys. 45, 150301 (2012)
J.Y. Choi, A stamp for all substrates. Nature 8, 311–312 (2013)
B.J. Cox, N. Thamwattana, J.M. Hill, Mechanics of atoms and fullerenes in single-walled carbon nanotubes. I. Acceptance and suction energies. Proc. R. Soc. Lond. Ser. A 463, 461 (2007)
B.J. Cox, N. Thamwattana, J.M. Hill, Mechanics of atoms and fullerenes in single-walled carbon nanotubes. II. Oscillatory behaviour. Proc. R. Soc. Lond. Ser. A 463, 477 (2007)
Y. Chan, N. Thamwattana, J.M. Hill, Axial buckling of multi-walled carbon nanotubes and nanopeapods. Eur. J. Mech. A Solid 30, 794–806 (2011)
X.Q. He, S. Kitipornchai, K.M. Liew, Buckling analysis of multi-walled carbon nanotubes: a continuum model accounting for van der waals interactions. J. Mech. Phys. Solids 53, 303–326 (2005)
D. Baowan, K. Chayantrakom, P. Satiracoo, B.J. Cox, Mathematical modelling for equilibrium configurations of concentric gold nanoparticles as potential application in drug and gene delivery. J. Math. Chem. 49, 1042–1053 (2011)
T.A. Hilder, J.M. Hill, Modeling the loading and unloading of drugs into nanotubes. Small 5, 300–308 (2009)
Y. Chan, J.M. Hill, Dynamics of benzene molecules situated in metal-organic frameworks. J. Math. Chem. 49, 2190–2209 (2011)
Y. Chan, J.M. Hill, Lithium ions storage between two graphene sheets. Nanoscale Res. Lett. 6, 203 (2011)
Y. Chan, J.M. Hill, Modelling interaction of atoms and ions with graphene. Micro Nano Lett. 5, 247–250 (2010)
Y. Chan, J.M. Hill, Hydrogen storage inside graphene-oxide frameworks. Nanotechnology 22, 305403 (2011)
A.W. Thornton, K.M. Nairn, J.M. Hill, A.J. Hill, M.R. Hill, Metal-organic frameworks impregnated with magnesium-decorated fullerenes for methane and hydrogen storage. J. Am. Chem. Soc. 131, 10662–10669 (2009)
W.X. Lim, A.W. Thornton, A.J. Hill, B.J. Cox, J.M. Hill, M.R. Hill, High performance hydrogen storage from Be-BTB metal-organic framework at room temperature. Langmuir 29, 8524–8533 (2013)
Y. Chan, R.K. Lee, J.M. Hill, Metallofullerenes in composite carbon nanotubes as a nanocomputing memory device. IEEE Trans. Nanotechnol. 10, 947–952 (2010)
Y. Chan, Mathematical modeling and simulations on massive hydrogen yield using functionalized nanomaterials. J. Math. Chem. 53, 1280–1293 (2015)
Y. Chan, L. Xia, Y. Ren, Y.-T. Chen, Multi-scale modelling on PM2.5 encapsulation inside doubly-layered graphene. Micro Nano Lett. 10, 696–699 (2015)
Y. Chan, J.M. Hill, Ion selectivity using membranes comprising functionalized carbon nanotubes. J. Math. Chem. 51, 1258–1273 (2013)
T. Pang, An Introduction to Computational Physics (Cambridge University Press, Cambridge, 2006)
J.E. Jones, On the determination of molecular fileds. I. From the variation of the viscosity of a gas with temperature. Proc. R. Soc. 106A, 441 (1924)
M. Sprik, M.L. Klein, K. Watanabe, Solvent polarization and hydration of the chlorine anion. J. Phys. Chem. 94, 6483–6488 (1990)
E. Weinan, Principles of Multiscale Modeling (Cambridge University Press, Cambridge, 2011)
G.C. Maitland, M. Rigby, E.B. Smith, W.A. Wakeham, Intermolecular Forces—Their Origin and Determination (Clarendon Press, Oxford, 1981)
Acknowledgments
We gratefully acknowledge the financial support from small research Grant (UNNC), and Ningbo Natural Science Foundation (2014A610025) and (2014A610172), and Qianjiang Talent Scheme (QJD1402009).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chan, Y. Modelling and MD simulations on ultra-filtration using graphene sheet. J Math Chem 54, 1041–1056 (2016). https://doi.org/10.1007/s10910-016-0606-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10910-016-0606-y