
Abstract
Wound care is a clinical challenge due to the susceptibility of the wound to multidrug-resistant bacterial infections. We report a multifunctional biocomposite of chitosan and gelatin (ratio = 1:1) based on silver nanoparticles (Ag NPs). The Ag NPs were selected due to their antimicrobial nature and were synthesised from an eco-friendly route using Ocimum sanctum (tulsi). The size of particles was 28.95 ± 7.74 nm in Transmission electron microscopy micrographs. Different concentrations of Ag NPs (0, 1, 2%, w/v) were used to prepare nano-biocomposite films (CG, CG1 and CG2) through the physical blending process followed by the solvent evaporation method. The physical properties of films, such as thickness and weight, were 0.10–0.12 mm and 0.93–1.55 g, respectively. Scanning electron microscopy and thermal gravimetric analysis studied the films' morphology and thermal stability. The Fourier transform infrared spectra affirmed the bond between silver and chitosan–gelatin. Nano-biocomposites showed high water uptake (300%) due to Ag NPs. Further, in vitro studies revealed antibacterial activity (Escherichia coli) with good in vitro biocompatibility and wound healing potential in the L929 mouse fibroblast cell line. Thus, the silver nanoparticle-biocomposite could be an effective topical dressing for biomedical applications.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Since ancient times, wound management has been a challenge for medical society due to its complexity. The major challenge during wound healing is the colonising bacteria or fungi leading to infection, which lowers growth hormones and destroys fibrin required for healing. Furthermore, bacteria trigger the inflammatory response; therefore, bacteria levels are frequently high in chronic wounds [1]. Antibiotic incorporated dressings are being administered to deal with these infections. Excessive use of antibiotics has led to the problem of bacterial resistance, demanding the urgent need for nanoparticles incorporated dressings [2,3,4]. Hence, 'nano-biocomposites' is the emerging field of nanomaterials for biomedical and tissue engineering. These composites have nanoparticles and biopolymers as fillers and matrices, respectively. These can be synthesised by the physical blending of components or in situ deposition of nanoparticles in the biopolymer matrix. Properties of composites depend on the nature and concentration of constituents and the nanoparticles' size and distribution [5].
Chitosan is preeminent and extensively used due to its hemostatic, biocompatibility, and non-toxic properties, such as in Hemcon®, Chitoflex® and Chitoseal™ [6]. Chitosan is the deacetylated form of chitin, a soluble cationic polysaccharide. The two units: 2-acetamido-2-deoxy-glucopyranose and 2-amino-2-deoxy-D-glucopyranose, are linked by β-(1 → 4). Therefore, it shows structural similarity with glycosaminoglycans. This component promotes bioactivity, cellular adhesion and activation of platelets and is one of the extracellular matrix (ECM) components [6, 7]. Chitosan's positively charged amino groups interact with erythrocytes' negatively charged cell membranes, thus preventing bleeding [8]. The other mode of chitosan for healing wounds is inducing vasospasm and mucoadhesion.
Chitosan-based dressings have already proven helpful in combat situations [9]. However, the neat chitosan is limited in processability, antimicrobial and mechanical properties [10]. To overcome these limitations, chitosan is modified by adding the crosslinker or using the chitosan composite in place of neat chitosan. Gelatin is a hygroscopic protein formed by partial hydrolysis of collagen [9]. This polymer has a film-forming capacity, wound healing property and prevents excess fluid loss from the wound site [11]. However, it lacks mechanical properties. Past studies have reported hemostatic dressings from the blend of chitosan and gelatin with improved thermal, mechanical properties, porosity and physicochemical properties compared to neat polymers [12]. These two biopolymers form a polyelectrolyte complex (PEC) by interacting an amino group of chitosan with the carboxyl group of gelatin [10, 13]. Li et al. prepared composite films of chitosan/gelatin loaded with ibuprofen [14]. Carvalho and Mansur developed hydrogels of chitosan–gelatin by UV radiation for chronic wounds [15]. Nguyen, Nguyen and Hsieh designed a sponge of chitosan/gelatin loaded with curcumin for wound healing applications [16]. The blend of chitosan and gelatin shows enhanced tensile strength and elongation [10]. Haung et al. evaluated the efficiency of carboxymethyl-chitosan/gelatin hydrogel for a full-thickness dermal wound model [17].
Moreover, the limited antimicrobial property of chitosan–gelatin can be improved by incorporating silver nanoparticles. Researchers are engaging silver nanoparticles (Ag NPs) for antimicrobial purposes in biomedical devices because excessive use of antibiotics resulted in increased drug resistant bacterial strains [18,19,20]. Ag NPs bind to the sulphur atom of the thiol group or the phosphorus atom present in the protein and DNA of the bacteria. Some scientists have even reported that particles attach to the cell wall disrupting the wall permeability and cellular respiration [21]. These NPs are also known for anti-inflammatory, antifungal, anti-platelet and anti-viral actions [22]. The nanoparticles are released slowly from the composites to inhibit the biofilms formed over the wound [4]. The plant-based synthesis of nanoparticles has many advantages, such as being non-toxic, cheap, and easy to prepare. It involves extract prepared from plants' parts, resulting in stable and biocompatible nanoparticles [3, 23]. Ocimum sanctum (tulsi) is a medicinal herb containing around 70% eugenol and 30% other phytochemicals (ursolic acid, carvacrol, linalool, camphene, tannin, etc.), which have potential to reduce and stabilise the nanoparticles [24,25,26].
Past studies have used green synthesis mediated silver nanoparticles to prepare films, hydrogels or dressings due to their biocompatibility, antimicrobial nature and large surface area to volume ratio for antimicrobial filtration, cancer treatment, food packaging and tissue engineering. Ediyilyam et al. and Kumar et al. prepared the silver nanoparticles incorporated biodegradable composite of chitosan and gelatin for food packaging films [27, 28]. Bhoir and Chawla reported antibacterial food packaging films of chitosan–gelatin having silver nanoparticles. These silver nanoparticles were synthesised using mint extract and polyvinyl alcohol as reducing and capping agents [29]. Paul, Jayan and Sasikumar prepared transdermal films with silver nanoparticles as the binding agent [30]. Venkatesan et al. developed a chitosan-alginate porous composite with biosynthesised silver nanoparticles for antimicrobial filtration and anticancer treatment [31]. Bardania et al. embedded green synthesised silver nanoparticles in polylactic acid/polyethylene glycol (PLA/PEG) film to develop antibacterial wound dressing [4].
The current study proposes incorporating eco-friendly, biocompatible and antibacterial Ag NPs prepared using O. sanctum in the PEC of chitosan and gelatin films for drug-resistant bacterial infection found in wounds which lead to more complications. As the crosslinker affects the biocompatibility of the PEC, no crosslinker was added in this study. The blending process is quite simple, cheap and greener. This study provides an insight into the improved physiochemical properties of nano-biocomposites and ascertains their efficacy as a promising topical wound healing dressing. The nano-biocomposites were tested against gram-negative bacteria (Escherichia coli) to show their antibacterial activity. The wound healing potential was examined by in vitro scratch assay using the L929 mouse fibroblast cell line.
2 Material and Methods
2.1 Materials
Chemicals including Silver nitrate (AgNO3, 99.9999% trace metal basis), chitosan (low molecular weight), gelatin, sodium hydroxide, glacial acetic acid, hydrochloric acid, 90 mm Bacterial culture dishes, Whatman filter paper, Luria Bertani broth, Dulbecco's Modified Eagle Medium (DMEM) were purchased from Sigma-Aldrich Chemicals Ltd., Germany. Glycerol, 3-(4,5-Dimethylthiazol2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), Calcein AM were purchased from Invitrogen, ThermoFisher Scientific, United States. Propidium iodide (PI) and foetal bovine serum (FBS) were purchased from Gibco, Life technologies, United States. Ocimum Sanctum (tulsi powder) and 96 & 24 well tissue culture plates were purchased from Swadesi, India and Corning, United States.
2.2 Preparation of Aqueous Extract of Tulsi Powder
The commercially available tulsi powder was used for extract preparation. The 1 g of tulsi powder was added to 100 mL of distilled water and boiled for 30 min while stirring in intervals. The extract was cooled, followed by filtration to obtain the filtrate, and then the filtrate was centrifuged in the cooling centrifuge at 7000 rpm for 20 min. Then the supernatant was collected and kept at 4 ºC for further use.
2.3 Synthesis of Ag NPs
2.3.1 Green Synthesis of Ag NPs
Silver nitrate (1 mM) solution was prepared and kept on heating at 60 ºC. Then, 5 mL of aqueous tulsi extract (1%) was added dropwise to 50 mL of silver nitrate under constant magnetic stirring. The colour change of the reaction from yellow to dark brown after 50 min confirmed the reduction of Ag+ to Ag0 [32]. The reaction mechanism has been explained in Fig. 1c. UV–Visible (UV–Vis) spectroscopy was used to monitor the reaction.

a Tulsi extract and b colloidal solution and c possible reaction mechanism of green synthesis of Ag NPs (Color figure online)
2.3.2 Purification of Ag NPs
The reaction mixture was transferred to oak ridge centrifuge tubes for centrifugation at 6000 rpm for 2 h, and then the supernatants were discarded. The pellets were washed 2–3 times with distilled water to remove the remaining unreacted extract and salt [33, 34]. The pellets were dried in a hot air oven at 40 °C for 7–8 h and stored at room temperature [32].
2.4 Characterisation of Ag NPs
UV–Vis spectroscopy is the preliminary characterisation technique used to monitor the synthesis of Ag NPs. Therefore, aliquots from the reaction mixtures were taken at different time intervals. The absorbance spectra were obtained by UV–Vis spectroscopy (Spectramax M2e, Molecular Devices) from the 300 to 700 nm wavelength range [18, 22, 35, 36]. The structure was analysed by powder X-ray diffraction (XRD) using monochromatic Cu–Kα radiation (λ = 1.5406 Ǻ), operating at 40 kV and 30 mA on a Bruker D8 Advance diffractometer. The scanning was done from 30º to 80º. The diffractograms were matched with the Joint Committee on Powder Diffraction Standards (JCPDS) library to study the crystallinity of particles [22, 36, 37]. The particle size was calculated by the well-known Debye Scherrer equation:
where, D = diameter of spherical particles, k = constant = 0.9, λ = wavelength of X-rays = 1.5406 Ǻ, β = full width half maxima (FWHM) corresponding to (111) plane and θ = Bragg’s angle.
The phytochemicals around the nanoparticles as stabilisers were determined by TGA 4000, PerkinElmer, when scanned over a temperature range from 20 to 600 ºC at a heating rate of 10 ºC/min under nitrogen gas flow [38]. Fourier Transform Infrared (FT-IR) spectroscopy help in identifying the functional groups of phytochemicals responsible for the reduction of silver ion (Ag+) present in silver nitrate (AgNO3) to silver (Ag0). Nanoparticles and the extract were analyzed by Perkin-Elmer Lambda 1050 spectrophotometer from the wavenumber of 400 cm−1 to 4000 cm−1 [22, 36, 37, 39, 40]. The synthesised nanoparticles were redispersed in water and then dried overnight under a vacuum on the copper grid to analyse the morphology of the particles using JEOL JEM-1400 Plus Transmission Electron Microscopy (TEM) operating at an accelerating voltage of 120 kV. The obtained images were studied with ImageJ software (National Institutes of Health and the Laboratory, USA) [35,36,37, 39]. The hydrodynamic diameter, polydispersity index and zeta potential were studied through the Malvern Zetasizer instrument [35, 41, 42].
2.5 Preparation of biocomposite films
Composite films were prepared by physical blending followed by solvent evaporation technique [28]. 2% (w/v) chitosan solution was prepared in 2% (v/v) of acetic solution using magnetic stirring at room temperature. Then, 2% (w/v) gelatin solution was prepared in distilled water using magnetic stirring at 40 ºC. Gelatin solution was added slowly to the chitosan solution in the ratio of 1:1 to obtain a polyelectrolyte complex using magnetic stirring for 1 h. 0.4% (v/v) of glycerol (as a plasticiser) was added to the composite using magnetic stirring for 1 h. Then, the film-forming solution was degassed, followed by Ultrabath sonication. Then, the 20 mL of solution was poured into the glass Petri dishes of 100 mm diameter and kept in a hot air oven at 40 °C for 7–8 h. Then films were peeled off using 1% (w/v) sodium hydroxide, washed with distilled water, then dried at room temperature and stored at 4 ºC for further use.
2.6 Preparation of Ag NPs-Based Biocomposite Films
The solution casting method is an applicable technique that provides good dispersion of the nano-fillers in the polymer or blend matrix. The 1 g and 2 g of Ag NPs were added to the 100 mL of two solutions of PEC, i.e., CG1 and CG2, respectively. These two solutions were kept under magnetic stirring for 30 min. After adding glycerol, the film-forming solution was degassed, followed by Ultrabath sonication. Then, the 20 mL of solution was poured into the glass Petri dishes of 100 mm diameter and kept in a hot air oven at 40 °C for 7–8 h. Then the films were peeled in similar ways, as mentioned in Sect. 2.5. The composition of each nano-biocomposite has been mentioned in Table 1.
2.7 Characterisation of Nano-biocomposites
2.7.1 Physical Properties
Weight was measured for each film (n = 3 for each formulation), and mean weight was calculated. The thickness of all the films was measured for uniformity by a screw gauge at six different locations, and then the average thickness was calculated [10, 27].
2.7.2 Water Uptake
Patches of the films (4 cm2) were immersed in 50 mL distilled water. After 0.5, 1, 1.5 and 24 h of immersion, excess water on the films was removed with blotting paper and then weighed. The water uptake (%) was calculated using the following equation:
where W and ware the weights of the films before and after immersion in the water, respectively [10, 27].
2.7.3 Dispersion Characteristics
The patches (4 cm2) of films were placed in 50 ± 1 mL of Phosphate buffer solution (pH 7.4) in a conical flask to determine the dispersion characteristics according to BS EN 13726–2:2001, Sect. 3.6. Then, the flask was swirled for 60 s, and after removing the patches from the flask, the dispersion was examined visually [43,44,45].
2.7.4 Stability Test
The solutions were sealed and stored at a refrigerated temperature of 5 ± 3 ºC for more than six weeks. The colour, appearance and texture were examined visually for sedimentation [11].
2.7.5 UV–Vis Spectroscopy
The absorbance of all the liquid samples of the films was recorded to confirm the presence of Ag NPs by using UV–Vis spectroscopy from the 300 to 700 nm range of wavelength [44].
2.7.6 Thermal Gravimetric Analysis (TGA)
The thermal stability of all films was carried out using TGA 4000, PerkinElmer. The chitosan, gelatin and composite films were scanned over a temperature range from 20 to 600 ºC at a heating rate of 10 ºC/min under nitrogen gas flow [10].
2.7.7 Attenuated Total Reflectance-Fourier Transform Infrared (FT-IR) Spectroscopy Analysis
The films were characterised by Spectrum Two spectrophotometer, PerkinElmer, to study the interaction among the components of the nano-biocomposites. The IR spectra were recorded in the wavenumber range of 400 cm−1 to 4000 cm−1 [10, 46].
2.7.8 Field Emission-Scanning Electron Microscope (FE-SEM) Imaging
The morphology and porosity of the films were imaged using the MIRA3 FE-SEM, Tescan. The films were sputter-coated with the gold particles and then observed at an accelerating voltage of 5 kV with a secondary electron image detector [46].
2.7.9 Transmission Electron Microscope (TEM)
The surface morphology and size of green synthesis mediated Ag NPs incorporated in chitosan–gelatin films were investigated by TEM (JEOL JEM-1400 Plus) at 120 kV. The sections of the samples were prepared using the diamond knife of cryotome (Lieca UC-6) at − 120 ºC with a thickness of 100 nm; then, the sections were placed on a copper grid and then dried under the lamp [31].
2.7.10 Antimicrobial Analysis
Multiresistant E. coli are associated with infections at surgical sites and burn injuries (skin and soft tissue infections). Therefore, E. coli was used to study the antimicrobial nature of nano-biocomposites. For disk diffusion assay, the composite solutions were placed on the Whatman Filter paper discs (5 mm) and then incubated with the bacterial culture for 24 h at 37 ºC. The diameters of the zone of inhibition around each disc were measured using a digital Vernier calliper [31].
For the contact killing method, an exponential growing bacterial culture was first diluted with PBS buffer. The samples (10 × 10 mm) were then loaded with 100 µL of bacterial suspension and cultured for 2 h at 37 °C. After the suspension was recovered, the samples were washed twice with 450 µL PBS. The removed bacterial suspension was mixed with the washing solution, and this solution was subjected to serial dilutions, then plated and incubated overnight at 37 °C for calculating colony forming units (CFU) [47, 48].
2.7.11 Cell Viability Studies
2.7.11.1 Cellular Viability Assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for assessment of cellular viability was performed on L929 mouse fibroblast cells, which were seeded in 96 well culture plates with a density of 5000 cells/well and incubated overnight at 37 °C in a 5% CO2 incubator. Treatments were given after 24 h of cell seeding. Further, MTT solutions (0.5 mg/mL) was added after 24 h of treatment, followed by 2 h of incubation in a CO2 incubator. After removing the medium, the Formazan crystals were dissolved in 200 µL of dimethyl sulphoxide (DMSO). On a Multiwell plate reader (Biotech Instruments, USA), absorbance was measured at 570 nm with a reference wavelength of 630 nm. The extract-based cytotoxicity assay for films was based on the ISO 10993-5 standard test for leachable products from medical devices. Here, the extract of three nano-biocomposite films was prepared by immersing the films in media (10 mg/mL) for 24 h [49,50,51]. The per cent cell viability was determined by using the equation,
2.7.11.2 Live/Dead Assay
Live/dead assay was done by CalceinAM/PI followed by fluorescence imaging to differentiate live and dead cells qualitatively for different concentrations of composite solutions (0.5, 1 and 2%). L929 cells were cultured in a 24-well culture plate with a density of 25,000 cells/well. The following day cells were treated with the composite solutions. After 24 h of incubation, the culture media was aspirated, and cells were washed twice with PBS, a 500 µL of Calcein-AM/Propidium iodide (PI) solution (3 µM Calcein AM and 5 µM PI in 5 mL 1× assay Buffer) was added. The plate was then incubated for 20 min in the dark and washed with PBS. 500 µL of culture medium was then added, and images (10X objective) were captured using an Inverted microscope of Olympus, Japan [51, 52].
2.7.12 In Vitro Scratch Assay
In vitro scratch assay is a facile, low-cost, and convenient, 2-dimensional method to evaluate cell migration [53]. The L929 fibroblast cells were seeded in 24-well plates (25,000 cells/well) and kept in a CO2 incubator for 24 h. A scratch was produced with a pipette's tip to trigger a wound and then washed with PBS. The wound was exposed to the treatment of composite solutions. The cells without treatment were used as the control. At 0 and 12 h after wounding, the digital images were taken with the Inverted microscope (×4 objective) to evaluate the cell migration rate [54, 55]. The wound area at 0 and 12 h was calculated using the wound healing size tool of ImageJ to measure the scratch closure [56].
where A0 and At are the wound area at time 0 and after 12 h, respectively.
2.8 Statistical Analysis
The quantitative results were expressed as mean ± standard deviation. Antimicrobial and in vitro scratch assay were analysed by one-way analysis of variance (ANOVA) with Tukey's post hoc test using Graphpad Prism software 8 (GraphPad Software, San Diego, California, USA).
3 Results
3.1 Characterisation of Plant-Mediated Ag NPs
3.1.1 Preliminary and UV–Vis Spectroscopy Studies
The brown colour's appearance affirmed the formation of Ag NPs in Fig. 1 because silver ions (Ag+) were reduced to silver (Ag) by the phytochemicals present in the plant extract. For this reaction, spectra at the different time points of 0, 10 and 50 min were recorded in Fig. 2a to monitor the completion rate.

a UV–Visible spectra at different time intervals, b XRD pattern, c TGA thermogram and d FT-IR spectra of Ag NPs
3.1.2 Surface Characterisation
XRD confirms the metal's presence, crystallinity, and structural information. The diffraction pattern in Fig. 2b showed 2θ peaks at 38.2°, 46.24°, 64.58° and 77.34o, which correspond to planes (111), (200), (220) and (311), respectively. These peaks were matched with the JCPDS (Joint Committee on Powder Diffraction Standards) file no. 04-0783 of the silver and confirmed the nanoparticles' fcc structure [57]. The particle size from the Debye Scherrer equation was 9.031 nm.
3.1.3 TGA Studies
From the TGA thermogram in Fig. 2c, the water loss was observed below 210 °C, and the weight loss from 210 to 550 °C was due to the desorption of phytochemicals surrounding the Ag NPs. It also showed that around 15% of the weight comprised phytochemicals (eugenol, ursolic acid, carvacrol, linalool, camphene, tannin) as capping agents around the nanoparticles [38, 58,59,60].
3.1.4 FT-IR Studies
This technique helps identify the functional groups of the phytochemicals responsible for reducing and stabilising the process. Therefore, the FT-IR of the extract and the green route Ag NPs were obtained (Fig. 2d). The following bands were found in the commercially available tulsi powder extract: 622.11, 711.81, 1074.61, 1115.00, 1267.18, 1402.15, 1605.71, 2935.40 and 3378.53 cm−1. The peak at 1074.61 and 1115.00 cm−1 for C–O–C stretching of ethers; 1267.18 cm−1 for C–O stretching of esters present in the flavones and terpenoids [61], 1402.15 cm−1 for S=O sulphate group of esters, 1605.71 cm−1 for C=O stretching of 3° amide present in proteins, 2935.40 cm−1 for C–H stretching in alkanes of lipids and 3378.53 cm−1 for stretching of O–H and N–H groups present in phenols and 3º amides respectively [35,36,37, 40]. The functional groups stabilising the silver nanoparticles showed a shift from 3378.53 to 3435.72 cm−1, 1402.15 to 1373.91 cm−1, 1267.18 to 1223.71 cm−1 and 1074.61 to 1028.45 cm−1. The shift in the C–O stretching implies involvement in the reduction of silver ions (Ag+) to silver (Ag0) [40].
From the above FT-IR spectral analysis, the possible mechanism of Ag NPs can be presumed (Fig. 1c). Eugenol gets oxidised to 4-allylidene-2-methoxy-cyclohexa-2,5-dienone while releasing e− and H+. Thus, Ag+ ions are reduced to Ag, further stabilised by O–H and C–O found in other phytochemicals [24,25,26, 62].
3.1.5 TEM Analysis
TEM micrograph of green synthesised Ag NPs is shown in Fig. 3a and b. Ag NPs were polydisperse and spherical shaped [35,36,37]. The mean particle size was 28.95 ± 7.74 nm by using Tulsi extract.

a TEM image and b the particle size distribution, c DLS and d Zeta potential of silver nanoparticles
3.1.6 DLS Analysis
The dynamic light scattering process is also known as elastic light scattering, which helps to determine the spherical particle's hydrodynamic diameter. The hydrodynamic diameter gives insight into the surface modification of the particles. The hydrodynamic diameter and zeta potential were 23.46 nm with a PDI of 0.626 and -17.8 mV, respectively, in Fig. 3c and d [41, 42, 63].
3.2 Characterisation of Nano-biocomposites
3.2.1 Physical Properties
Neat chitosan films are difficult to remove from Petri-dishes due to being brittle; therefore, researchers have employed a blend of chitosan–gelatin [64]. The developing nano-biocomposite from chitosan–gelatin (ratio = 1:1) is explained by schematic Fig. 4. In preliminary observation, all films were transparent, marginally brittle and homogenous. The only difference found was the appearance of reddish-brown colour in CG1 and CG2 films than CG due to the incorporation of silver nanoparticles (Fig. 5a). The physical properties have been mentioned in Table 1 and shown in Fig. 5b and c. The weight and thickness of films ranged from 0.93 to 1.55 g and 0.10 to 0.12 mm respectively. Hence, uniformly thick films were fabricated by the solvent evaporation method.

Schematic representation of the preparation of biocomposites

a Image, b mean thickness, c weight, and d water uptake (%) of films (Color figure online)
3.2.2 Water Uptake
The water uptake capacity of a wound healing dressing is significantly essential to enhance its role in fast healing by absorbing excess wound exudate. Figure 5d showed that the weight of films increased for an initial one hour when kept in water, and after that, no more water was absorbed. Hence, CG1 could be proposed as a potential dressing for wound management.
3.2.3 Dispersion Characteristics
The dispersion characteristics determine the change in physical properties of a wound dressing when it interacts with the wound fluid. Here, the result revealed the retention of the original structure, which implies that these films will remove from the wound site without causing any pain [43,44,45].
3.2.4 Stability Test
No sedimentation or aggregation was found in all three composite solutions after six weeks, which indicated the stability of the colloidal solution. The chitosan–gelatin solution had a clear solution, while the other nano-biocomposites (CG1 and CG2) solutions retained the colour of Ag NPs [11].
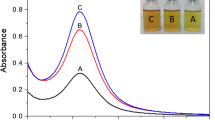
3.2.5 UV–Visible Spectroscopy
The solutions of all nano-biocomposites were analysed by this spectroscopy (Fig. 6a). Standalone composite (CG) did not show any absorbance in the UV–Vis spectrum, while nano-biocomposites (CG1 and CG2) showed absorbance at 435 nm, indicating the presence of Ag NPs. The absorbance increased with the increasing concentration of Ag NPs in the composite from 1 to 2% [27, 31, 65].

a UV–Vis spectra, b TGA thermogram of chitosan (C), gelatin (CG), and chitosan–gelatin (CG), c TGA thermogram of chitosan–gelatin (CG), nano-biocomposites (CG1 and CG2) and d FT-IR spectra of nano-biocomposites
3.2.6 TGA Studies
The thermal stability of chitosan (C), gelatin (G),chitosan–gelatin (CG) and chitosan–gelatin-Ag NPs (CG1 and CG2) were studied and illustrated in Fig. 6b and c. Weight loss was measured at 45–100 ºC, 100–420 ºC, 420–500 ºC and 520–650 ºC. The weight loss of films up to 100 ºC was due to water evaporation. Weight loss from 100 to 420 ºC is due to the decomposition of amine and –CH2OH of Chitosan. Pyrolysis of gelatin occurred from 420 to 500 ºC. Then, rings of chitosan decomposed up to 600 ºC. The chitosan–gelatin (CG) blend showed similar decomposition ranges, but the remaining weight was higher than the individual biopolymers [10, 11].
3.2.7 FT-IR Studies
The FT-IR spectra of nano-biocomposites are exhibited in Fig. 6d. The band at 3295 cm-1 is due to the stretching vibrations of O–H and N–H groups of chitosan and gelatin [10, 11, 27, 28]. The bands at 2930 and 2876 cm-1 are attributed to asymmetric vibration of –C–H [11, 28], 1647 cm−1 for stretching vibration of –C=O (amide I), 1550 cm−1 is due to bending vibration of –N–H (amide II) [27, 28, 66]. The bands at 1452 cm−1 assigned to bending vibration of –CH2 [10, 11], 1402 cm−1 for –COO− [10, 67], 1237 cm−1 for –C≡N vibration (amide III) [66], 1032 cm−1 is due to stretching vibration of C–O–C, and 923 and 850 cm−1 is assigned to cyclohexane ring vibrations[15]. The results obtained for the CG film is consistent with those previously published in the literature.
The incorporation of Ag NPs in nano-biocomposites increased the intensities of bands at 2876, 1647 and 1237 cm−1 in the IR spectra [27, 28, 68, 69].
3.2.8 FE-SEM Analysis
The surface morphology of the Chitosan–gelatin films with or without Ag NPs is shown in Fig. 7a. In SEM micrographs, the NPs on the surface of films was visible. Hence, the surface of CG films was smooth, homogeneous and compact, while that of films with Ag NPs were coarse and heterogeneous [27, 28].

a SEM and bTEM micrographs of films
3.2.9 TEM Analysis
The TEM images of nano-biocomposite (CG1 and CG2) is shown Fig. 7b. Micrographs showed that a dense polymeric shell surrounded the Ag NPs. The size of the Ag NPs did not change even after blending with chitosan–gelatin [31].
3.2.10 Antimicrobial Analysis
The antimicrobial results for the nano-biocomposite solutions with E. coli were demonstrated in Fig. 8a and b. For disk diffusion assay, the chitosan–gelatin (CG) composite blend showed marginal inhibitory activity against E.coli. The zone of inhibition was 7.46 ± 0.50 and 9.68 ± 0.77 mm for CG1 and CG2, respectively, against 7.00 ± 0.29 mm for CG. Therefore, the increasing content of Ag NPs in biocomposites improved the antimicrobial activity [31]. The above results are supported by the results of the contact killing method (Fig. S1).

a Zone of inhibition and b Antimicrobial activity of nano-biocomposite against E. coli, ***p < 0.001
3.2.11 Cell Viability Studies
3.2.11.1 MTT Assay
The extraction-based method examines the cytotoxicity of Ag+ ions released from films when kept in the simulated wound environment. The toxicity of solution and extract media is illustrated in Fig. 9a and b. A lower concentration of Ag was released from CG1 compared to CG2. These NPs are capped by the phytochemicals that may bind with the amino groups of proteins of cells, ultimately reducing the toxic nature of CG1. Hence, CG1 is safer than CG and CG2 extracts [70].

Cell viability studies: MTT assay a direct, b extract and c Fluorescent images of live/dead assay calcein AM (green), and propidium iodide (red) represents live and dead cells, respectively (Color figure online)
3.2.11.2 Live/Dead Assay
Further cytotoxicity analysis assisted by CalceinAM/PI fluorescence imaging assay showed all formulations to be safe at all screened concentrations (Fig. 9c). No significant dead population was seen [71].
3.2.12 In Vitro Scratch Assay
The scratch assay is an accessible method for assessing the migration of the epidermal layer. It evaluated the healing process on treatment with nano-biocomposite for 12 h exposure in Fig. 10a and b. The fibroblast exposed to CG closed the scratch by about 40% compared to the control. The CG1 treatment significantly closed the scratch wounds by about 45%. So, the closure rate was highest in CG1 compared to other treatments.

a Phase-contrast images of the wound at 0 and 12 hour. b Bar graph for the qualitative results of in vitro scratch assay. *p < 0.05, **p < 0.01 and ***p < 0.001
4 Discussions
With the rise of drug-resistant bacterial infections, multifunctional biomaterials that can treat acute and post-operative wounds are becoming increasingly important. In this study, the principal finding is that the silver nanoparticles-based biocomposite possesses properties such as preventing infection and increasing wound closure rate. It can be a good candidate for topical dressing required for wound care.
The synthesis of nanoparticles is already well established. In our study, we have used biological extract to limit the use of toxic solvents because of its application as a wound healing dressing. Past studies have used different parts of plants such as leaves, roots, seeds, peel, and flowers for plant-mediated synthesis. These studies have reported the antibacterial activity of Ag NPs ranging in size from 3 to 100 nm [72]. Ramteke et al. prepared spherical Ag NPs of 18 nm from an aqueous extract of O. sanctum leaves [73]. Singhal et al. prepared bio-reduced Ag NPs of size 4–30 nm within 8 min of reaction time. Ag Nps exhibited more antibacterial activity against gram-negative (Escherichia coli) and gram-positive (Staphylococcus aureus) bacteria than ciprofloxacin and silver nitrate [41]. Here, we have used commercially available tulsi powder due to its rich composition of eugenol and other phytochemicals. Because of these phytochemicals, tulsi shows the following properties: antioxidant, anti-inflammatory, immuno-modulator and antibacterial. The Ag NPs were further added to the blend of polymers. The AgNPs gave an absorbance at 429 nm in UV–Vis spectra [40]. The structure of Ag NPs was face-centred cubic from the XRD pattern. The extra peaks (*) were due to the crystallinity of the phytochemicals present on the surface of Ag NPs [36, 57, 74, 75]. TGA studies also confirmed the presence of phytochemicals as capping agents on Ag NPs [24, 25]. In colloidal solution, the size of spherical shaped Ag NPs ranged from 20 to 35 nm. Biocompatible Ag NPs can be employed for a wide range of applications, including wound healing, drug delivery, medical diagnosis & devices, biosensing, health care, water purification, and the food industry [28]. The polydispersity of nanoparticles limits the application of green synthesis in different fields.
Many therapies have been used to prevent infection, and encourage wound healing. Dressings based on silver nanoparticles have been used as a quick and efficient wound healing therapy to deal with growing bacterial resistance. Past studies have reported using Ag NPs loaded blend biopolymers or PEC for food packaging, cancer treatment and wound management. When loaded with the drug, PEC can maintain swelling and release profiles of the drug. Ediyilyam et al. prepared Ag NPs from Mussaenda frondosa, showing an absorption peak at 426 nm. The size of nanoparticles was from 10 to 30 nm. Green synthesised nanoparticles were incorporated into the PEC of chitosan and gelatin (ratio = 9:1). The spread plate method evaluated the PEC films for food packaging. The film containing AgNPs lowered the bacterial contamination compared to plastic polyethene films on the seventh day of storage [27]. Kumar et al. used aqueous fresh fruit extract of Mimusops elengi to prepare Ag NPs. The absorbance peak was found at 427 nm, and the size ranged from 25 to 45 nm. This research group first added Ag NPs to gelatin solution, then added this to chitosan solution in the ratio of (Chitosan:gelatin = 9:1) for food packaging purposes. The films were wrapped around fresh grapes for shelf-life study [28]. Bhoir et al. synthesised PVA capped AgNPs by mixing aqueous poly vinyl alcohol, mint extract, and silver nitrate solutions. The theoretical radius of NPs with and without capping agent was 7.8 nm and 8.8 nm, respectively. The chitosan–gelatin films were incorporated with Ag NPs for food packaging application. These films showed better water vapour transmission rate and mechanical properties than the pure blend [29].
In contrast, we have used Ag NPs to prepare biocompatible films for wound care management. As far as we know, only one research work has been published on incorporating Ag NPs in chitosan–gelatin transdermal films. Paul et al. synthesised colloidal silver from an aqueous extract of Ganoderma lucidum. In the UV–Vis spectrum of colloidal silver, the absorbance peak at 418 nm was found. Spherical shaped Ag NPs was of 10–50 nm. The films were prepared by the solution casting method. In this study, the physicochemical properties of nanoparticle incorporated films were not compared with the neat films of chitosan–gelatin. The transdermal films were examined by biological studies such as disc diffusion assays and MTT assay. However, these films contained the unreacted extract of Ganoderma lucidum, which might dominate antibacterial activity and toxicity results compared to the loading of purified Ag NPs [30].
The present work prepared the films by the physical blending process, devoid of any crosslinker. The different concentrations of purified silver nanoparticles ranged from 0 to 2% (w/v) incorporated in the PEC of chitosan–gelatin. In UV–Vis spectra of composite solutions, absorbance at 435 nm is due to the presence of silver in the solution. The slight shift in absorbance might be due to the aggregation of Ag NPs while preparing the nano-biocomposite. The SEM images also further confirmed the aggregation of AgNPs on the surface of films. This aggregation did not affect the wound healing properties. This incorporation of silver nanoparticles improved the physicochemical properties of the films [27, 28]. It was noticed in Fig. 5b that with decreased content of polymers, thickness decreased. From Fig. 5d, CG1 showed the best water holding capacity. The hydrophilicity of AgNPs based on biocomposites is due to water-soluble phytochemicals on the surface of nanoparticles [27, 76]. The formation of coordination bonds between the blend of chitosan–gelatin led to increased bands' intensities at 2876, 1647 and 1237 cm−1 in the IR spectra [27, 28, 68, 69]. From Fig. 6c, it was deduced that about 22% of the weight remained at 600 ºC, indicating the stronger interaction between chitosan and gelatin (CG). About 10% of CG1 and CG2 weight remained at 600 ºC due to the coordination bond between Ag NPs and the blend. As a result, absence of the free amino group of chitosan and the carboxyl group of gelatin for interaction between the two polymers in nano-biocomposite. The increased concentration of Ag NPs from CG1 to CG2 led to the slight decrease in the remaining weight. The results of CG1 and CG2 in TGA are in good agreement with the FT-IR studies, highlighting the bond formed between the blend of chitosan–gelatin and Ag NPs.
Multidrug-resistant E. coli is of major concern due to its association with skin and soft tissue. E.coli was used for disk diffusion assay (Fig. 8) and contact killing method (Fig. S1). In blend, the polyelectrolyte complex of chitosan and gelatin resulted in less freely available amino groups of chitosan, which interacts with the negatively charged bacterial cell wall, leading to disruption of the cellular functions. In contrast, CG1 and CG2 showed leaching of Ag NPs. Here, CG2 showed a statistically significant zone of inhibitions against E. coli. The antibacterial property of Ag NPs is well known for disrupting the DNA and stimulating oxidative stress, ultimately inducing bacterial death. Companion tests examining the cytotoxic effects of soluble silver concentrations provided crucial insight into the compatibility of the nano-biocomposites employed in our study. The presence of biomolecules on the surface of NPs considerably alters particle-cell interactions; yet, the relationship between green synthesis mediated silver nanoparticles and cellular absorption, and cytotoxicity is still poorly understood [70]. In extract-based MTT assay, the cells treated with CG1 showed higher cell viability than CG and CG2 up to 40% of extract media. In the direct composite solution-MTT assay, CG and CG1 were safe up to the concentration of 2.5%, while CG2 showed slight toxicity at the same concentration. Although Ag NPs have benefits in therapeutics, few pieces of research have emphasised the toxic nature of Ag NPs. It is crucial to interrogate the live/dead cells after incubating with composite solutions. There were no changes in cell morphologies when cultured directly with different concentrations of composite solutions (0.5, 1 and 2%). When the cells were exposed to effective concentration (2%) of CG1, then spread, and spindle-like cells were found to be attached on the culture plate. After all, most cells were still viable in all the groups [71]. Hence, the results of all cell viability studies have highlighted the safety of CG1 films. These nano-biocomposites (CG1 and CG2) are safe for post-operative and skin wounds due to their eco-friendly synthesis route.
Cell proliferation and migration are critical aspects of tissue formation in wound healing. Damage to the epithelial monolayer causes an increase in growth factors and cytokines, which causes cells to migrate and proliferate, forming a robust intercellular adhesion at the wound site. Past studies have already pointed out the wound healing capacity of chitosan and gelatin. Therefore, these two polymers are being used in tissue engineering. Here, our study has also confirmed the wound healing property of CG due to the reasonable cell migration rate. Interestingly, CG1 treated 2-D wound showed a significantly improved cell migration across the scratch in 12 h compared to control, CG and CG2 in Fig. 9a and b. The lower wound closure for CG2 treatment might be due to the slight toxicity with increased Ag NPs content in the nano-biocomposite. The above result favours the application of CG1 for wound healing dressing [54, 55]. These results also emphasise that these biocompatible Ag NPs are capped by different phytochemicals, which might also act as a catalyst in speeding up the wound healing process, as reported by past research [77, 78]. Therefore, these nano-biocomposites hold many advantages over the composites containing chemically synthesised Ag NPs.
With the above favourable results from the current study, it is reasonable that green synthesis mediated Ag NPs loaded biocomposites films with high water uptake capacity, noticeable antibacterial activity, and promoting cell proliferation could be favourable for faster healing of acute and surgical wounds. Even then, more future studies are required to have clear insight into the implications of the application of topical films in animal models.
5 Conclusion
Biopolymer films containing green synthesised silver nanoparticles (1 or 2%) are simple and fast to manufacture, making them commercially appealing. Biocompatible and spherical Ag NPs of 20–35 nm was used as an antibacterial drug. Ag NPs in biocomposite was confirmed by UV Vis spectroscopy and TEM images. At the same time, FT-IR and TGA results confirmed the bond formation between the blend of chitosan–gelatin and Ag NPs. These results firmly acknowledged this nano-biocomposite. The water holding capacity of the films improved with the addition of Ag NPs, which is an essential factor for developing wound healing dressings. The cell viability studies such as MTT and Live/Dead assay have shown non-toxic nature on L929 cells. The studies were completed by in vitro scratch assay on L929 cell lines for 12 h. This assay seemed to distinguish between composites in terms of their potential to facilitate wound healing by promoting fibroblast cell migration. Therefore, all of the above studies showed noticeable results for applying green synthesised silver nanoparticle-based biocomposites as an antibacterial wound dressing for acute and post-operative wounds.
Data Availability
All data generated or analysed during this study are included in this published article.
References
M.E. Okur, I.D. Karantas, Z. Şenyiğit, N. Üstündağ Okur, P.I. Siafaka, Asian J. Pharm. Sci. 15, 661 (2020)
D.M. Suflet, I. Popescu, I.M. Pelin, D.L. Ichim, O.M. Daraba, M. Constantin, G. Fundueanu, Pharmaceutics 13, 1461 (2021)
R. Augustine, A. Hasan, J. Drug Deliv. Sci. Technol. 56, 101516 (2020)
H. Bardania, R. Mahmoudi, H. Bagheri, Z. Salehpour, M.H. Fouani, B. Darabian, S.S. Khoramrooz, A. Mousavizadeh, M. Kowsari, S.E. Moosavifard, G. Christiansen, D. Javeshghani, M. Alipour, M. Akrami, Sci. Rep. 10, 1 (2020)
X. Wang, J. Chang, C. Wu, Appl. Mater. Today 11, 308 (2018)
W. Paul and C. P. Sharma, Adv. Wound Heal. Mater. (2015).
S. Stratton, N.B. Shelke, K. Hoshino, S. Rudraiah, S.G. Kumbar, Bioact. Mater. 1, 93 (2016)
H.T. Peng, Mil. Med. Res. 7, 13 (2020)
N. R. Kunio and M. A. Schreiber, in Consult. Hemost. Thromb., Third Edit (Elsevier, 2013), pp. 538–545.
N. Nady, S.H. Kandil, Membranes (Basel). 8, 2 (2018)
S. Murali, S. Kumar, J. Koh, S. Seena, P. Singh, A. Ramalho, A.J.F.N. Sobral, Cellulose 26, 5347 (2019)
M.A. Khan, M. Mujahid, Int. J. Biol. Macromol. 124, 138 (2019)
M. Bin Ahmad, J.J. Lim, K. Shameli, N.A. Ibrahim, M.Y. Tay, Molecules 16, 7237 (2011)
H. Li, F. Cheng, S. Gao, Z. Wu, L. Dong, S. Lin, Z. Luo, X. Li, J. Appl. Polym. Sci. 134, 45441 (2017)
I.C. Carvalho, H.S. Mansur, Mater. Sci. Eng. C 78, 690 (2017)
V.C. Nguyen, V.B. Nguyen, M.-F. Hsieh, Int. J. Polym. Sci. 2013, 1 (2013)
X. Huang, Y. Zhang, X. Zhang, L. Xu, X. Chen, S. Wei, Mater. Sci. Eng. C 33, 4816 (2013)
S. Pirtarighat, M. Ghannadnia, S. Baghshahi, J. Nanostructure Chem. 9, 1 (2019)
M. Pandima Devi, Int. J. Pharma Bio Sci. 7, P38 (2016)
G. Sathishkumar, C. Gobinath, K. Karpagam, V. Hemamalini, K. Premkumar, S. Sivaramakrishnan, Colloids Surfaces B Biointerfaces 95, 235 (2012)
A.D. Russell, W.B. Hugo, Prog. Med. Chem. 31, 351–370 (1994)
N. Krithiga, A. Rajalakshmi, A. Jayachitra, J. Nanosci. 2015, 1 (2015)
N.K. Rajendran, S.S.D. Kumar, N.N. Houreld, H. Abrahamse, J. Drug Deliv. Sci. Technol. 44, 421 (2018)
S. Zafar, A. Zafar, Open Biotechnol. J. 13, 37 (2019)
S.B. Aziz, G. Hussein, M.A. Brza, S.J. Mohammed, R.T. Abdulwahid, S. Raza Saeed, A. Hassanzadeh, Nanomaterials 9, 1557 (2019)
A. Usmani, P.P. Dash, A. Mishra, Mater. Sci Adv. Compos. Mater. 2, 1 (2018)
S. Ediyilyam, B. George, S.S. Shankar, T.T. Dennis, S. Wacławek, M. Černík, V.V.T. Padil, Polymers (Basel). 13, 1680 (2021)
S. Kumar, A. Shukla, P.P. Baul, A. Mitra, D. Halder, Food Packag. Shelf Life 16, 178 (2018)
S.A. Bhoir, S.P. Chawla, Int. J. Nanosci. 16, 1650022 (2017)
S. Paul, A. Jayan, C.S. Sasikumar, Asian Pacific J. Trop. Dis. 5, 975 (2015)
J. Venkatesan, J.-Y. Lee, D.S. Kang, S. Anil, S.-K. Kim, M.S. Shim, D.G. Kim, Int. J. Biol. Macromol. 98, 515 (2017)
K. Paulkumar, G. Gnanajobitha, M. Vanaja, M. Pavunraj, G. Annadurai, Adv. Nat. Sci. Nanosci. Nanotechnol. 8, 035019 (2017)
E. Parthiban, N. Manivannan, R. Ramanibai, N. Mathivanan, Biotechnol. Rep. 21, e00297 (2019)
J. S. Moodley, S. B. N. Krishna, K. Pillay, Sershen, and P. Govender, Adv. Nat. Sci. Nanosci. Nanotechnol. 9, 015011 (2018).
D. Bharathi, D.M. Josebin, S. Vasantharaj, V. Bhuvaneshwari, J. Nanostruct. Chem. 8, 83 (2018)
K. Anandalakshmi, J. Venugobal, V. Ramasamy, Appl. Nanosci. 6, 399 (2016)
K. Shameli, M. Ahmad, P. Shabanzadeh, A. Zamanian, P. Sangpour, Y. Abdollahi, Z. Mohsen, Int. J. Nanomedicine 7, 5603 (2012)
D. Parida, C. Bakkali-Hassani, E. Lebraud, C. Schatz, S. Grelier, D. Taton, J. Vignolle, Nanoscale 14, 4635 (2022)
D. Nguyen, J. Lee, K. Park, Y. Ching, X. Nguyen, V.H. Phan, T. Hoang Thi, Nanomaterials 10, 542 (2020)
O. Erdogan, M. Abbak, G.M. Demirbolat, F. Birtekocak, M. Aksel, S. Pasa, O. Cevik, PLoS ONE 14, e0216496 (2019)
G. Singhal, R. Bhavesh, K. Kasariya, A.R. Sharma, R.P. Singh, J. Nanoparticle Res. 13, 2981 (2011)
Z. A. Ali, R. Yahya, S. D. Sekaran, and R. Puteh, Adv. Mater. Sci. Eng. 2016, (2016).
S. Hasatsri, A. Pitiratanaworanat, S. Swangwit, C. Boochakul, C. Tragoonsupachai, Dermatol. Res. Pract. 2018, 1 (2018)
R. Tandon, S. Gandhi, S. Khushu, R.P. Tripathi, J. Chitin Chitosan Sci. 2, 35 (2014)
M. Uzun, S.C. Anand, T. Shah, J. Biomed. Eng. Technol. 1, 1 (2013)
D. Tripathi, A. Sharma, P. Tyagi, C.S. Beniwal, G. Mittal, A. Jamini, H. Singh, A. Tyagi, AAPS PharmSciTech 22, 1 (2021)
R. Nazir, D. Parida, J. Borgstädt, S. Lehner, M. Jovic, D. Rentsch, E. Bülbül, A. Huch, S. Altenried, Q. Ren, P. Rupper, S. Annaheim, S. Gaan, Chem. Eng. J. 417, 128028 (2021)
A.V. Fuchs, S. Ritz, S. Pütz, V. Mailänder, K. Landfester, U. Ziener, Biomater. Sci. 1, 470 (2013)
K. Nešporová, V. Pavlík, B. Šafránková, H. Vágnerová, P. Odráška, O. Žídek, N. Císařová, S. Skoroplyas, L. Kubala, V. Velebný, Sci. Rep. 10, 1 (2020)
B. Boonkaew, P. Suwanpreuksa, L. Cuttle, P. M. Barber, and P. Supaphol, J. Appl. Polym. Sci. 131 (2014).
M.O. Wang, J.M. Etheridge, J.A. Thompson, C.E. Vorwald, D. Dean, J.P. Fisher, Biomacromol 14, 1321 (2013)
D. Zhang, Z. Hu, L. Zhang, S. Lu, F. Liang, S. Li, Materials (Basel). 13, 5038 (2020)
C.C. Liang, A.Y. Park, J.L. Guan, Nat. Protoc. 2, 329 (2007)
G. Yaşayan, G. Karaca, Z.P. Akgüner, A. Bal Öztürk, Int. J. Polym. Mater. Polym. Biomater. 70, 623 (2021)
K. Ravishankar, M. Venkatesan, R.P. Desingh, A. Mahalingam, B. Sadhasivam, R. Subramaniyam, R. Dhamodharan, Mater. Sci. Eng. C 102, 447 (2019)
A. Suarez-Arnedo, F. Torres-Figueroa, C. Clavijo, P. Arbeláez, J.C. Cruz, C. Muñoz-Camargo, PLoS ONE 15, e0232565 (2020)
M. Behravan, A. Hossein Panahi, A. Naghizadeh, M. Ziaee, R. Mahdavi, A. Mirzapour, Int. J. Biol. Macromol. 124, 148 (2019)
B. Adebayo Tayo, A. Salaam, A. Ajibade, Heliyon 5, e02502 (2019)
M.M.H. Khalil, E.H. Ismail, K.Z. El-Baghdady, D. Mohamed, Arab. J. Chem. 7, 1131 (2014)
Annu, A. Ali, and S. Ahmed, Handb. Ecomater. 1 (2018).
S. Ahmed, Saifullah, M. Ahmad, B. L. Swami, and S. Ikram, J. Radiat. Res. Appl. Sci. 9, 1 (2016).
J. Singh, T. Dutta, K.H. Kim, M. Rawat, P. Samddar, P. Kumar, J. Nanobiotechnology 16, 1 (2018)
M. Sundrarajan, S. Ambika, K. Bharathi, Adv. Powder Technol. 26, 1294 (2015)
S. Patel, S. Srivastava, M.R. Singh, D. Singh, Int. J. Biol. Macromol. 107, 1888 (2018)
W. Cao, J. Yan, C. Liu, J. Zhang, H. Wang, X. Gao, H. Yan, B. Niu, W. Li, Carbohydr. Polym. 247, 116643 (2020)
S. Ahmed, S. Ikram, J. Photochem. Photobiol. B Biol. 163, 115 (2016)
R.A. Mauricio-Sánchez, R. Salazar, J.G. Luna-Bárcenas, A. Mendoza-Galván, Vib. Spectrosc. 94, 1 (2018)
L. Ding, X. Shan, X. Zhao, H. Zha, X. Chen, J. Wang, C. Cai, X. Wang, G. Li, J. Hao, G. Yu, Carbohydr. Polym. 157, 1538 (2017)
V.Q. Nguyen, M. Ishihara, J. Kinoda, H. Hattori, S. Nakamura, T. Ono, Y. Miyahira, T. Matsui, J. Nanobiotechnology 12, 1 (2014)
A. Regiel-Futyra, M. Kus-Liśkiewicz, V. Sebastian, S. Irusta, M. Arruebo, A. Kyzioł, G. Stochel, RSC Adv. 7, 52398 (2017)
S.R. ur Rehman, R. Augustine, A.A. Zahid, R. Ahmed, M. Tariq, A. Hasan, Int. J. Nanomedicine 14, 9603 (2019)
V. Holubnycha, O. Kalinkevich, O. Ivashchenko, M. Pogorielov, Nanoscale Res. Lett. 13, 1 (2018)
C. Ramteke, T. Chakrabarti, B.K. Sarangi, R. Pandey, J. Chem. 2013, 1 (2013)
S. Jain, M.S. Mehata, Sci. Rep. 7, 1 (2017)
Y. Rout, J. Microbiol. Antimicrob. 4, 103 (2012)
A.M. Díez-Pascual, A.L. Díez-Vicente, Biomacromol 16, 2631 (2015)
S. Shetty, S. Udupa, L. Udupa, Evidence-Based Complement. Altern. Med. 5, 95 (2008)
R. Barreto, R. Albuquerque-Júnior, A. Araújo, J. Almeida, M. Santos, A. Barreto, J. DeSantana, P. Siqueira-Lima, J. Quintans, L. Quintans-Júnior, Molecules 19, 846 (2014)
Acknowledgements
The authors are grateful to the Director, Institute of Nuclear Medicine and Allied Sciences, Defence Research and Development Organisation, Delhi, India and Indian Institute of Technology, Delhi, and India for financial support. Isha Gupta would like to acknowledge the Union Grant Commission, New Delhi, India, for providing fellowship (Fellowship No. 114562). Abhishek Kumar is a fellowship recipient from the Council of Scientific and Industrial Research, New Delhi, India.
Funding
Funding was provided by University Grants Commission (Grant No.: 114562) and Council of Scientific and Industrial Research, India
Author information
Authors and Affiliations
Contributions
IG: Conceptualisation, data curation, formal analysis, writing, methodology, original draft preparation, review and editing. AK: Data curation, analysis. ANB: Resources, review and editing. SS: Resources, supervision, review and editing. SG: Conceptualisation, funding acquisition, review and editing, resources, supervision, project administration.
Corresponding author
Ethics declarations
Conflict of interest
The authors testify that they have no conflict of interest in the subject matter discussed in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gupta, I., Kumar, A., Bhatt, A.N. et al. Green Synthesis-Mediated Silver Nanoparticles Based Biocomposite Films for Wound Healing Application. J Inorg Organomet Polym 32, 2994–3011 (2022). https://doi.org/10.1007/s10904-022-02333-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-022-02333-w