
Abstract
Europium doped yttrium fluoride (YF3:Eu) nanoparticles(core-NPs) with and without a passive layer of LaF3 coating around the core-NPs were prepared by simple polyol process. The surface of these luminescent core/shell-NPs was additionally functionalized with amorphous silica. The phase purity, crystalinity, shapes and size of the as-prepared core and their respective core/shell-NPs were determined by X-ray diffraction (XRD) pattern and transmission electron microscopy (TEM). The as-prepared core/shell-NPs were successfully grafted with silica layer as verified by Fourier transform infrared spectroscopy (FTIR). As observed from absorption spectra, the energy band gap was decreased after core/shell formation because of increase in grain size of the luminescent-NPs. The core/shell-nanostructure synthesized by this method exhibited a remarkable enhancement than their respective core-NPs. However, the emission intensity was suppressed after silica surface coating because of non-radiative transitions effect. The presence of silanol (Si–OH) groups on their surfaces not only results in high solubility in aqueous solvents, but also allows further conjugation with biomolecules such as antigens, antibodies and oligonucleotides, which pave the way for potential bio-applications of the core/shell/SiO2-NPs.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In recent years, considerable efforts have been devoted to the synthesis of luminescent NPs of lanthanide because of their sharp absorption and emission transitions in visible region, high quantum yield, long life time, superior photo-stability, high chemical and thermal stability, excellent biocompatibility and low toxicity which make them essential candidates for their potential use in various field of applied material as well as in biomedical sciences [1–5]. In comparison to various kind of traditional oxide based luminescent lanthanide materials such as Ln2O3, [6, 7] LnPO4 [8, 9] and LnVO4 (where Ln = Y, La and Gd) [10–12] fluorides are advantageous as fluorescent host materials owing to their low vibrational energies and the subsequent minimization of the quenching of the excited state of the lanthanide ions [13–23]. Hence, lanthanide ions-doped nanostructural fluorides have attracted extensive interests due to their potential applications in lighting, displays, up-converters, magnetic resonance imaging (MRI), biological fluorescent labels, optical amplifiers, transparent glass, scintillators, photonic crystals [13–23], etc. Among these fluorides, YF3 host matrix exhibits high photochemical stability, excellent biocompatibility, non-toxicity and relatively low crystalline temperature, and its phonon energy is as low as 350 cm−1 [14–18, 21–23]. The lanthanide ions (Eu3+, Tb3+, Er3+, Pr3+, Ho3+, Yb3+, Tm3+) doped YF3 NPs, YF3 triangular nanoplates and silica-coated or organic ligands modified YF3 luminescent NPs have been reported in literatures [15–18, 21–25]. The luminescent quantum yield of NPs is limited due to the non-radiative decay from surface defects. In order to increase the quantum yield, a good way is to form a core/shell structure [26–33]. This core/shell structure consists of a core doped with luminescent lanthanide ion which is covered by shells of similar lattice constant materials. In this structure the distance between the luminescent lanthanide ions and the surface quenchers is increased, thus, reducing the nonradiative pathways. In these core/shell materials design, the outer shell protects the luminescent core ions from oxidation, hydrolysis or dissolving in aqueous environment at room temperature and decreases their solubility in most of the solvents. Owing to the surface coating of an inert crystalline layer, these luminescent NPs revealed weak solubility in aqueous and non-aqueous solvents. Therefore, their applications in biomedical sciences are limited because they don’t have additional functional chemical groups for attachment of biomolecules.
To overcome this problem, it is better to grow a porous silica shell surrounding the core/shell NPs, since the silica shell is easy to grow on the surface of luminescent core-NPs and has the ability to enhance the solubility, bio-compatibly and non-toxic nature of the luminescent core/shell-NPs [14, 19, 26, 30, 34, 35]. Silica coating is a popular method for improving water solubility and protecting NP cores from the surrounding environment [19, 26, 30, 34, 35]. Inexpensive and easy method of obtaining spherical particles with narrow size distributions, chemical inertness and optical transparency are some of the advantages of using SiO2 as shell in NPs. Increased optical response, higher luminescence and longer decay times have been observed when the cores of luminescent Ln3+-doped materials are covered by SiO2 shells [19, 26, 30, 34, 35]. Because of the low chemical activity of SiO2, the coating process seems to be an effective way of lowering the cytotoxicity of the cores. However, no one has reported such core/shell/SiO2-nanostructure and also no research have been done to investigate their structural, optical and photoluminescence properties.
In the present study, we propose a facile and effective strategy to grow a new kind of core/shell/SiO2 nano-struture to enhance the luminescent efficiency along with their solubility in aqueous and non-aqueous solvents. The obtained core-NPs exhibited bright orange/red emission which was remarkably enhanced after passive LaF3 layer coating and then luminescent intensity got suppressed due to the amorphous silica surface coating, although, their solubility character in aqueous and non-aqueous solvents was improved significantly. The existence of the silanol (Si–OH) groups on the surface of core/shell-NPs is an advantageous over unmodified cores. These groups could be used for chemical modifications and conjugation of biomolecules such as biotin, antibodies and oligonucleotides. The silica shell also improves the water solubility of the NPs, and is known as an anti-agglomerating agent. Therefore, it is important to study the effect of surface coatings on the spectroscopic properties of NP cores. The aim of this research was to investigate the effect of surface coating on structural, optical and photoluminescence properties of luminescent seed core-NPs. This work is still under investigation to evaluate the cyto-toxicity, biocompatibility and their further use in biological sciences.
2 Materials and Methods
Yttrium oxide (99%, BDH chemicals Ltd, England), Europium oxide (99.99%, Alfa Aesar, Germany), ethanol (E-Merck, Germany), Tetraethyl orthosilicate (TEOS, 99 wt% analytical reagent A.R.),ethylene glycol(EG), NH4F, HNO3 and NH4OH were used as the starting materials without any further purification. Y(NO3)36H2O and Eu(NO3)36H2O were prepared by dissolving the corresponding oxides in diluted nitric acid. The de-ionized water was prepared using a Milli-Q system (Millipore, Bedford, MA, USA). All other chemicals used were of reagent grade.
For preparation of Eu3+-doped YF3 NPs (Y0.95Eu0.05F3), 0.2 M stock solutions of Y(NO3)36H2O and Eu(NO3)36H2O in de-ionized water were prepared. In brief, 9.5 ml of Y(NO3)36H2O and 0.5 ml of Eu(NO3)36H2O were dissolved in 50 ml of ethylene glycol. Then an equiv. molar aqueous solution of NH4F was added drop wise under magnetically stirred forgoing reaction, and kept whole solution with magnetic stirring on hotplate at 80 °C to obtain homogenously mixing. Later, this homogenously mixed solution was transferred in a round bottle 250 ml flask fitted with reflux condenser and waited for 4 h until complete precipitation. On cooling to room temperature, the white precipitates segregated to the bottom. The product was collected by centrifugation and washed with distilled water and absolute ethanol several times, and dried in oven at 60 °C for 6 h for further characterization. The obtained solid product can be re-dispersed in de-ionized water to form a water-dispersible solution.
For the preparation of YF3:Eu3+@LaF3 core/shell NPs, similar polyol process was used as discussed above. The as-prepared 0.500 g YF3:Eu3+ were dispersed with the help of ultra-sonication in 10 ml of distilled water. These dispersed NPs solution mixed into magnetically stirred hot EG dissolved La(NO3)36H2O (0.500 g) solution. After thirty minutes an equiv. molar aqueous solution of NH4F was injected into the foregoing mixed system under magnetic stirring at 80 °C. Afterward this suspension was refluxed at 80 °C for 3 h until the complete precipitation is occurred. This white precipitate was centrifuged and washed many times with ethanol and dist. water to remove excess un-reacted reactants. The core/shell NPs were collected after centrifugation and allowed to dry in ambient temperature for further characterization.
The YF3:Eu3+@LaF3@SiO2 core/shell NPs were prepared through a versatile solution sol–gel method as follows [19, 26, 30, 34, 35]. The synthesized YF3:Eu3+@LaF3 NPs (50 mg) were well dispersed in a mixed solution of deionized water (50 mL), ethanol (70 mL) and concentrated aqueous ammonia (1.0 mL) in a three-neck round-bottom flask. Afterward, 1.0 mL of tetraethyl orthosilicate (TEOS) was added drop-wise in 2 min, and the reaction was allowed to proceed for 5–6 h under continuous mechanical stirring. After continuous stirring at room temperature, the silica-coated YF3:Eu3+@LaF3 core/shell NPs were separated by centrifugation, washed several times with ethanol and dried at room temperature for further analysis.
The crystalinity of the powder samples was examined by X-ray diffraction (XRD) at room temperature with the use of Rigaku X-ray diffractometer equipped with a Ni filter using Cu Kα (λ = 1.54056 Å) radiations as X-ray source. The size and morphology of the samples were inspected using a field emission transmission electron microscope (FE-TEM) equipped with the EDX (FETEM, JEM-2100F, JEOL, Japan) operating at an accelerating voltage of 200 kV. EDX analysis was used to confirm the presence of the elements. The samples for TEM were prepared by depositing a drop of a colloidal ethanol solution of the powder sample onto a carbon-coated copper grid. The FTIR spectra were recorded on a Perkin-Elmer 580B IR spectrometer using KBr pellet technique in the range 4000–400 cm− 1. The UV/Vis absorption spectra were measured in the Perkin-Elmer Lambda-40 spectrophotometer in the range 190–600 nm, with the sample contained in 1 cm3 stoppered quartz cell of 1 cm path length. Photoluminescence spectra were recorded on Fluorolog 3 spectrometer model: FL3-11, Horiba JobinYvon Edison MJ USA. All measurements were performed at room temperature.
3 Results and Discussion
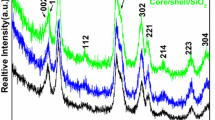
The crystal structure, phase analysis and crystalinity of the prepared products were verified from X-ray diffraction pattern. As seen in Fig. 1, all three samples exhibit the all reflection planes of well crystalline cubic YF3 (space group Pnma(62)) cell parameters (a = 6.35, b = 6.85, c = 4.39 A), which are in good agreement with the standard data (JCPDS card No. 072–0579) [15–17], although some impurities of orthorhombic phase (JCPDS No. 074–0911) are also present in the sample. Absence of any other impurity peak suggests that luminescent Eu3+ ion has been effectively doped into the YF3 lattice site and pure crystalline phase of orthorhombic YF3:Eu could be obtained from this facile synthesis rout. It is worth noticing that, all reflection peaks are broad that may be result from nanocrystalline size of the building units, lattice imperfections and stacking faults in the contacting area between neighboring particles in the structure. Although the reflection peak positions are same, relative peaks intensity corresponding to (101), (131) and (231) planes are varied significantly in the observed core–shell pattern (Fig. 1 (core)). We believe that it could be due to the possible tropism of the crystals and probable anisotropy growth of shell. Furthermore, enhancement in peak intensity could be related to increase in the grain size and similar structure between YF3 and LaF3. Moreover, it is difficult to distinguish the diffraction planes of LaF3 shell just from the XRD pattern. We believe that YF3 and LaF3 are iso-structural (cubic phase) crystalline materials and similar lattice constant of these two materials, resulting the XRD pattern not to change after LaF3 layer deposition. It is apparent in Fig. 1 (core/shell), that diffraction peak intensities are decreased owing to the amorphous silica shell growth on the surface of core/shell NPs. Average grain sizes of the samples estimated according to the Scherrer formula from the most strongest peak (2θ= 44°) are to be 12, 17 and 22 nm, for core, core/shell and core/shell/SiO2 NPs. The increases of acrystaline size suggest the growth of shell on the surface of core-NPs.

X-ray diffraction pattern of core, core/shell and core/shell/SiO2 NPs
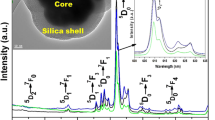
Transmission electron microscopy was carried out to examine the morphology, micro-structural details and surface coating of silica around the core/shell NPs. As observed in Fig. 2a, luminescent core-NPs are irregular grain shaped particles which are highly aggregated with narrow size distribution, with average particle size of the core-NPs between 10 and 15 nm supported by XRD results through Debye–Scherrer formula. In this micrographs dark area with highly aggregated small particles are related to luminescent core-NPs. However, light grey color smooth amorphous silica shell is surrounding the aggregated luminescent core-NPs. It is mentioning that the presence of hydrated (EG) groups on the surface of as-prepared luminescent core-NPs enhanced their aggregation in aqueous solvent. These results are in good agreement with a previous report by Yan et al., who synthesized different Ln3+ ions doped YF3 nanocrystals in aqueous environment [15–17, 35]. Previously some researchers have observed that EG promotes increase in aggregation process of small particles or nucleation seeds on the surface through the lateral interaction of hydrogen bonding of H2O with the –OH groups of this ethylene glycol [19–23, 33, 36]. Silica surface modification is performed over aggregated core/shell-NPs and the particles are still aggregated with narrow size distribution [16, 19, 35, 36]. But these core/shell nanostructures are considerably larger in size ~40 nm than the parent luminescent core-NPs. This is because amorphous silica shell has been effectively encapsulated surrounding the core/shell-NPs through so-gel process as seen in Fig. 2a, b. An approximately 15 nm thick silica shell is coated over aggregated core/shell nanostructures. Because of hydrated surface and high aggregation of core-NPs, amorphous silica can easily react with the surface of core-NPs and form thick layer surrounding the aggregated core-NPs. EDX analysis further supported this hypothesis. Energy dispersive X-ray analysis was carried out to determine the doping, chemical composition and atomic ratio in the as-prepared nano-product. The EDX results in Fig. 2c, display that the core/shell/SiO2-NPs are mainly composed of Y, La, F, O and Si elements. Note that the strong signals for C and Cu come from the carbon coated copper TEM grid. No other impurity peak can be detected, supporting the XRD results.

FE-TEM images of (a and b) core and core/shell/SiO2 NPs (c) energy dispersive X-ray analysis of core–shell-SiO2 NPs
FTIR spectroscopy was performed to further confirm the grafting of silica shell around the core/shell NPs and to understand the surface chemistry before surface coating of nanocrystals. Notably, a strong doublet strong peak at around 1150 cm−1 and relatively sharp intensity peaks at 951 and 799 cm−1are observed, which correspond to the symmetrical stretching and bending vibrations of (Si–O), (Si–O–Si) and (Si–OH) groups from the surface anchored amorphous silica [19, 26, 34, 36]. All three samples display broad infrared absorption band located at 3436 cm−1 along with weak intensity bands at around 1620 and 1430 cm−1, which originate from symmetrical stretching, bending and wagging vibration modes of (O–H) groups from surface bound physically adsorbed residual water molecules [19, 26, 34, 36]. The infrared absorption band observed at around 475 cm− 1 is attributed to the absorption of M-O network. Since these samples are prepared in aqueous solvents, we expect that their surface may be covered by a large amount of (O–H) groups either chemically attached or physically adsorbed on the surface of NPs. Additionally, crystalline water may be adsorbed onto the surface of luminescent NPs as a coordinating ligand with lanthanide ions. These observations are in good agreement with the literature reports [8]. These surface hydroxyl groups increase the hydrophilicity of the materials in aqueous solvents (Figs. 3, 4).

FTIR spectra of the core, core–shell and core–shell-SiO2 NPs

a UV–Vis absorption spectra of core, core/shell and core/shell/SiO2 NPs in de-ionized water and in inset shows the plot of (αhν)2 vs. photon energy (hν) of the core, core/shell and core/shell/SiO2 NPs. b UV–vis absorption spectra of core, core–shell and core–shell-SiO2 NPs in absolute ethanol and in inset shows the plot of (αhν)2 vs. photon energy (hν) of the core, core/shell and core/shell/SiO2 NPs
Optical properties and colloidal stability were determined by UV–visible spectroscopy. Figure 4 illustrates the absorption spectra of core, core/shell and core/shell/SiO2 NPs in dist. water over the range from 200 to 600 nm under UV–vis region. From Fig. 4a, a significant enhancement is observed in absorption spectrum of silica surface modified NPs in respect to non-modified core/shell NPs since amorphous silica surface coating enhanced dispersibility of NPs in aqueous solvent. The high colloidal dispersibility of surface modified core/shell/SiO2 NPs in dist. water as well as in ethanol suggests that optically active amorphous silica layer has been successfully encapsulated around the surface of core/shell NPs. Moreover, non-modified NPs also show dispersibility in aqueous solvent, as they are prepared in aqueous solvent and hydroxyl groups are covered around the surface of NPs either chemically bonded or physically adsorbed on the surface of NPs. We consider that these surface hydrophilic (hydroxyl) groups may increase their dispersibility in aqueous solvents. The content of hydroxyl groups on the surface of core, core/shell NPs are also determined by FTIR spectroscopy. We observed similar trend in absorption spectra measured in ethanol as shown in Fig. 4b. According to the Tauc and Menth law the experimentally estimated band gap energies of core, core/shell and core/shell/SiO2-NPs are found to be 2.14, 2.13 and 2.07 eV in H2O and 1.98, 1.97 and 1.86 eV in absolute ethanol, respectively [37]. We believe that, the discrepancies in the band gap energies could be related to the grain size. As the grain size increases after surface coating the energy band gap decreases. The decrease in the band gap can be attributed to defects, local bond distortion, intrinsic surface states, and interfaces that yield localized electronic levels in the forbidden band gap. We believe that this significant difference is attributed to surface and interface intrinsic defects and quantum confinement effects linked to the nano-crystalinity.

Excitation spectra of the core, core–shell and core–shell-SiO2 NPs
In order to examine the effect of surface coating, we observed the photoluminescence spectra of core, core/shell and core/shell/SiO2-NPs at room temperature. Figure 5 displays the excitation spectra of core and their respective inert LaF3 and silica surface coated core/shell-NPs by monitoring the emission at 593 nm corresponding to the 5D0→7F1 transition. As shown in Fig. 5, all observed excitation spectrum composed of many sharp excitation transitions in UV/Vis region. The observed sharp excitation peaks in near UV region consisting at 316 (7F0→5H3,6), 360 (7F0→5D4), 370–385 (7F0→5G3), 393 (7F0→5L6), 413 (7F0→5D3) and 463 (7F0→5D2), which attributed to intra-configurational parity forbidden 4f 6-4f 6 excitation electronic transitions originating from 7F0 ground state to the labeled excited states of Eu3+ ions [1, 38–40]. These observed excitation transitions of Eu3+ ions are agreeing well with the published literature reports [38–40]. Figure 6 illustrates the comparative emission spectra of core, core/shell and core/shell/SiO2-NPs excited using 393 nm under similar experimental conditions within the spectral range from 400 to 750 nm. The emission spectra of all three samples composed of three strong and three weak emission transitions in visible region assigned to 523–535 (5D1→7F1), 553 (5D1→7F2), 585–593 (5D0→7F1), 608–619 (5D0→7F2), 641–650 (5D0→7F3), and 686–700 (5D1→5F4), deriving from intra-configurational parity forbidden 4f 6-4f 6 -excitation electronic transitions originating from 5D0 ground state to the labeled excited states of Eu3+ ions [1, 5, 11]. Among the recorded emission transitions, magnetically dipole allowed transition observed in visible region located at 595 nm (5D0→7F1) is most prominent transition in all three samples because of similar crystal structure. And the intensity of (5D0→7F1) transition is much stronger than that of the (5D0→7F2) transition, indicating good mono-chromaticity and the inversion symmetry of the Eu3+ site. As seen in Fig. 6, after surface modification no notable shift in peak positions of the emission transitions is occurred since the 4f energy levels of Eu3+ are hardly affected by the crystal field because of the shielding effect of the 5s25p6 electrons. But the selection rules and transition probabilities depend on the crystal field. It is well-known that the 5D0 →7F2 transition is parity forbidden and can be observed only when the lattice environment is distorted and contains a noninversion symmetry [1]. Being a forced electric dipole transition, this transition is hypersensitive to the environment. However, the 5D0→7F1 transition is insensitive to the environment due to allowed magnetic dipole consideration and the ratio of the two intensities is a good measure for the symmetry of the Eu3+ site. The 5D0→7F1 magnetic dipole transition in the inversion symmetry site is dominating, while the 5D0→7F2 electric dipole transition in the noninversion symmetry site is the strongest. In the emission spectrum of as-prepared YF3:Eu3+ nanocrystals, the most intense peaks are centered at 585 and 591 nm. Both peaks correspond to the magnetic dipole transitions (5D0→7F1) and much stronger than the 610 and 619 nm (5D0→7F2) emission. Therefore, it is inferred that the Eu3+ ions in YF3 located a site with inversion symmetry. The other transition 5D0→7F3 has a mixed electric dipole and magnetic dipole character while transition 5D0→7F4 is an electric dipole transition.

Emission spectra of the core, core–shell and core–shell-SiO2 NPs
On comparing the emission spectra of three samples, the relative peak intensity is little stronger in core/shell NPs compared to core-NPs. The increase intensity could have resulted from the increase in size of the core-NPs after surface growth of similar passive crystal structure LaF3 shell. Moreover, growth of inert LaF3 shell around the core-NPs protects the doped luminescent lanthanide ions from non-radiative transitions, which originate from high vibration energies or other quenching sites located at the surface of the NPs. It is known that the emission intensity of luminescent lanthanides ions will be quenched to some extent in the environment that has a high phonon frequency such as –OH groups which have high vibrational frequency near 3450 cm−1, also confirmed by FTIR results [28–36]. Thus epitaxial growth of passive LaF3 layer effectively reduces non-radiative quenching of the luminescent lanthanide ions by surface defects and surrounding surface adsorbed organic moieties. Furthermore, considerable enhancement in luminescence efficiency is attributed to the facts that the LaF3 layer has been grafted successfully, which protect the luminescent core-NPs from non-radiative transitions. Subsequently, we grafted an amorphous silica layer around core/shell NPs through the hydrolysis and condensation of silanol groups. However, after coating a silica layer luminescent intensity is suppressed to some extent because of the light scattering effect on both emission and incident light by the porous silica layer. This phenomenon can be attributed to the involvement of large vibrational modes, such as Si–OH on the surface of NPs, which may cause considerable non-radiative transition and reduce the luminescence intensity of luminescent lanthanide ions. A lot of papers have also reported that non-radiative centers such as –OH group exist on the surface [26, 28, 33, 36] and they proved that the silica surface modified NPs could suppress the luminescent intensities because that the silica shell network amplify the surface defects. On the other hand, these luminescent quenching silanol molecules greatly enhanced the solubility, colloidal stability, biocompatibility and nontoxic nature of the as-prepared NCs. The amorphous silica coating was conducted in our work for further biological applications and to maintain luminescent efficiency.
4 Conclusions
In summary, well crystallized YF3:Eu, YF3:Eu@LaF3 and silica surface modified YF3:Eu@LaF3@SiO2-NPs were prepared successfully. These core and core/shell/SiO2-NPs ranging from 35 to 40 nm are well dispersed in aqueous solvent as verified from UV/Vis spectra. FTIR analysis confirmed the silica surface modification, resulting the successful formation of YF3:Eu@LaF3@SiO2 core/shell nano-structure. The decrease in experimentally estimated energy band gap from core to core/shell/SiO2-NPs is attributed to the quantum size effect because growth of passive LaF3 and silica layers enhanced the grain size of the luminescent core-NPs. The core/shell-NPs revealed enhanced luminescent intensity due to the shielding effect, in which formation of passive LaF3 layer surrounding the luminescent core-NPs protect from incident and emission light scattering. The silica modified core/shell/SiO2-NPs shows lower emission intensity because of absorption of UV exciting radiation by the SiO2 shell and its quenching properties. These results suggest safety and adequacy of NPs for future use in medical or biological applications. Covering NPs by SiO2 shell opens up new possibilities like modification of the surface of core/shell structures or chemical connection with important organic molecules/drugs. This understanding may facilitate synthesis of orange/red nanophosphors with a biocompatible coating suitable for display and bioimaging applications.
References
F.S. Richardson, Chem. Rev. 82, 541 (1982)
P. Huhtinen, M. Kivela, O. Kuronen, V. Hagren, H. Takalo, H. Tenhu, T. Lovgren, H. Harma, Anal. Chem. 77, 2643 (2005)
Z. Chen, W. Zheng, P. Huang, D. Tu, S. Zhou, M. Huang, X. Chen, Nanoscale 7, 4274 (2015)
I. Hemmila, V. Laitala, J. Fluoresc. 15, 529 (2005)
A.A. Ansari, T.N. Hasan, N.A. Syed, J.P. Labis, A.K. Parchur, G. Shafi, A.A. Alshatwi, Nanomedicine 9, 1328 (2013)
S. Som, S.K. Sharma, J. Phys. D 45, 415102 (2012)
T.S. Atabaev, H.K. Kim, Y.H. Hwang, J. Colloid Interface Sci. 373, 14 (2012)
M.N. Luwang, R.S. Ningthoujam, Jagannath, S.K. Srivastava, R.K. Vatsa, J. Am. Chem. Soc. 132, 2759 (2010)
A.K. Gulnar, V. Sudarsan, R.K. Vatsa, T. Sakuntala, A.K. Tyagi, U.K. Gautamd, A. Vinu, Nanoscale 2, 2847 (2010)
G. Wang, W. Qin, D. Zhang, L. Wang, G. Wei, P. Zhu, R. Kim, J. Phys. Chem. C 112(44), 17042 (2008)
A.A. Ansari, J.P. Labis, Mater. Lett. 88, 152 (2012)
M. Yu, J. Lin, J. Fang, Chem. Mater. 17, 1783 (2005)
Z.L. Wang, Z.W. Quan, P.Y. Jia, C.K. Lin, Y. Luo, Y. Chen, J. Fang, W. Zhou, C.J. O’Connor, J. Lin, Chem. Mater. 18, 2030 (2006)
A.A. Ansari, S.P. Singh, N. Singh, B.D. Malhotra, Spectrochim. Acta. Part A 86, 432 (2012)
R. Yan, Y. Li, Adv. Mater. 15, 763 (2005)
S. Sarkar, V. Mahalingam, Cryst. Eng. Comm. 15, 5750 (2013)
S.L. Zhong, Y. Lu, M.R. Gao, S.J. Liu, J. Peng, L.C. Zhang, S.H. Yu, Chem. Eur. J. 18, 5222 (2012)
C. Peng, C. Li, G. Li, S. Li, J. Lin, Dalton Trans. 41, 8660 (2012)
S. Sivakumar, P.R. Diamente, F.C.J.M. van Veggel, Chem. Eur. J. 12, 5878 (2006)
F. Wang, Y. Zhang, X. Fan, M. Wang, J. Mater. Chem. 16, 1031 (2006)
G. Liu, X. Li, X. Dong, J. Wang, J. Nanopart. Res. 13, 4025 (2011)
N.O. Nunez, M. Ocana, Nanotechnology 18, 455606 (2007)
L. Wang, Y. Zhang, Y. Zhu, Nano Res. 3, 317 (2010)
F. Pelle, M. Dhaouadi, L. Michely, P. Aschehoug, A. Toncelli, S. Veronesi, M. Tonelli, Phys. Chem. Chem. Phys. 13, 17453 (2011)
G. Chen, H. Qiu, R. Fan, S. Hao, S. Tan, C. Yang, G. Han, J. Mater. Chem. 22, 20190 (2012)
A.A. Ansari, R. Yadav, S.B. Rai, RSC Adv. 6(26), 22074 (2016)
F. Vetrone, R. Naccache, V. Mahalingam, C.G. Morgan, J.A. Capobianco, Adv. Funct. Mater. 19, 2924 (2009)
A.K. Parchur, A.I. Prasad, A.A. Ansari, S.B. Rai, R.S. Ningthoujam, Dalton Trans. 41, 11032 (2012)
J.C. Boyer, J. Gagnon, L.A. Cuccia, J.A. Capobianco, Chem. Mater 19, 3358 (2007)
A.A. Ansari, A.K. Parchur, M. Alam, J. Labis, A. Azzeer, J. Fluoresc. 24, 1253 (2014)
A. Kar, S. Kundu, A. Patra, Chem. Phys. Chem. 16, 505 (2015)
K. Kompe, H. Borchert, J. Storz, A. Lobo, S. Adam, T. Moller, M. Haase, Angew. Chem. Int. Ed. 42, 5513 (2003)
G.S. Yi, G.M. Chow, Chem. Mater. 19, 341 (2007)
A.A. Ansari, M. Alam, J.P. Labis, S.A. Alrokayan, G. Shafi, T.N. Hasan, A.A. Alshatwi, J. Mater. Chem. 21, 19310 (2011)
M. Darbandi, T. Nann, Chem. Commun. 776 (2006)
K. Dabrowska, M. Giersig, S. Lis, T. Grzyb, M. Runowski, J. Nanopart. Res. 15, 1958 (2013)
J. Tauc, A. Menth, J. Non-Cryst. Solids 8–9, 569 (1972)
S.K. Gupta, P.S. Ghosh, M. Sahu, K. Bhattacharyy, R. Tewari, V. Natarajan, RSC Adv. 5, 58832 (2015)
T.J. deS. Ramos, R. daS. Viana, L. Schaidhauer, T. Cassol, S.A. Junior, J. Mater. Chem. C 3, 10934 (2015)
J. Legendziewicz, V. Tsaryuk, V. Zolin, E. Lebedeva, M.M. Borzechowskaa, M. Karbowiak, New J. Chem. 25, 1037 (2001)
Acknowledgements
This work was supported through the project funded by national plan for science, technology and innovation (MAARIFAH) King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award Number (13-Bio1246-02).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ansari, A.A., Manthrammel, M.A. Surface Coating Effect on Structural, Optical and Photoluminescence Properties of Eu3+ Doped Yttrium Fluoride Nanoparticles. J Inorg Organomet Polym 27, 194–200 (2017). https://doi.org/10.1007/s10904-016-0463-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-016-0463-y