
Abstract
The influence of carbon and silicon atoms doping on the structural and electronic properties of the (BeO)12 nanocluster is investigated through density functional theory calculations. It has been found that doping process induces local deformation at bond lengths and angles near the doping site. The results indicate that C or Si doping decreases the energy gap of the (BeO)12 nanocluster. It seems that the electronic character of the (BeO)12 nanocluster could be adjusted by particular impurity. The electronic charge distributions are also analyzed using Atoms in Molecules theory. Natural bond orbital analyses are also performed for scrutinizing the structural properties of the considered nanoclusters.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Discovery of carbon nanotube [1] causes a fundamental revolution in material science and technology. After that, nanoscale materials have attracted considerable interest due to their unique properties and extreme different functionalities. Recently exploring new nanoscale materials are of growing interest, because of their exotic properties. Moreover many attempts have been made to investigate inorganic-based nanostructures which exhibit somewhat different properties [2–6]. Among different kinds of nanostructures, II-VI inorganic ones are attracted extreme interest due to their unique electronic and magnetic properties [7, 8]. Beryllium oxide nanotubes were proposed and their structural, cohesive, and electronic properties were predicted [9–12]. Because of great ionic character of Be–O bond, beryllium oxide compounds often have different properties from their C, BN and SiC counterparts. For example, wurtzite BeO is an insulator with wide band gap, high melting point, high thermal conductivity and large elastic constants [13]. These advantages distinguish the BeO nanostructures as considerable potential for applications in nanoelectronic devices. Besides, beryllium and beryllium oxides are valuable materials in the atomic energy, rockets, missiles, aviations, polymer chemistry and metalluragical industry.
It is noteworthy that investigating fullerene-like structures has become one of the most important topics in nanotechnology, because of their important applications in electronic devices, imaging materials, magnetic recording and environmental processes [11, 14–22]. Recently, Ren et al. [23] studied the structure of (BeO)N clusters using the method combining the genetic algorithm with density function theory. They reported that the global minimum structures of (BeO)N clusters are cages at N ≥ 8, among which (BeO)12 is one of the most viable cluster. Moreover it was shown that (BeO)12 has a fullerene-like structure as well as it was indicated that the bonding pattern includes two kinds of σ-bonds (one belongs to four-membered rings, and another belongs to six-membered rings) through adaptive natural density partitioning (AdNDP) analysis. It is noticeable that the fullerene-like clusters (XY)12 have been theoretically predicted to be the most viable clusters among different types of (XY)n structures [23–26]. On the other hand, research on the doped nanostructures is a hot spot for scientists. For instance, Ryzhkov et al. [27] reported a magnetization of the wurzite-like beryllium oxide induced by carbon doping by means of the density functional theory. Also it was shown that magnetism in BeO nanotubes is induced in the presence of carbon impurities [28]. Therefore it would be interesting to investigate the impact of the carbon and silicon doping on the structural and electronic properties of (BeO)12 cluster. The main contribution of this work is to study the influence of carbon and silicon doping on the different properties of (BeO)12 through density functional theory (DFT) calculations. It should be mentioned that the experimental measurement of the physical properties of (BeO)12 is not explored yet. However these hypothetical doped non-doped (BeO)12 nanoclusters exist only in silico.
2 Computational Details
The geometries of all considered structures are fully optimized using B3LYP method [29, 30] and 6 − 311 + G* [31, 32] basis set and the nature of the stationary points is checked by frequency analysis at the same computational level of theory. It is noteworthy that pure DFT methods underestimate HOMO–LUMO gaps relative to hybrid DFT methods such as B3LYP that include Hartree–Fock exchange function. DFT hybrid methods lead to significant improvement of the gaps due to the cancellation of different errors [33, 34]. All calculations are performed using Gaussian 03 package [35]. The Atoms in Molecules (AIM) analysis for the obtained electronic wave functions is done using AIM2000 suite of program [36]. Moreover the electronic properties of the considered structures are investigated through their density of states (DOS) spectra obtained from GaussSum program [37].
The natural bond orbital (NBO) analysis is also performed in the present study. According to the NBO a two-center \({{\sigma }}_{\rm{AB}}\) bond NBO between atoms A and B is defined as
where hA, hB are the respective hybrids on the atoms, and \(c_{A}\), \(c_{B}\) are the natural polarization coefficients. The polarity of each \({{\sigma }}_{\text{AB}}\) bond could be quantified in terms of the natural ionicity parameter \(i_{AB}\), fined as
The natural ionicity \(i_{AB}\)is zero for a pure covalent bond (\(c_{A}^{{}} = c_{B}^{{}}\)) but can achieve any value between −1 (\(c_{A}^{{}} = 0\); pure ionic hybrid on B) and +1 (\(c_{B}^{{}} = 0\); pure ionic hybrid on A), ranging smoothly between ionic and covalent limits [38]. Moreover, the Wiberg bond index (WBI) [39], which is a measure of bond character, is also considered. The WBI comes from the manipulation of the density matrix in the orthogonal natural atomic orbital based on the NBO analysis and is defined as
in which\(p_{jk}\)is the density matrix element and therefore \(p_{jj}\)simply denotes the charge density in the jth atomic orbital. This index closely relates to the bond order, the larger WBI implies to stronger covalent character. All NBO calculations are performed through NBO 5.G implemented in Gaussian 03 package.
Recently, many attempts have been made to introduce new concepts and principles which can be used in describing the reactivity, selectivity or stability of molecular systems. Among these concepts energy gaps (\(E_{g}\)) and hardness (\(\eta\)) values are considered to explore electronic properties of the considered clusters. Using Janak’s approximation [40] these quantities have the following operational equation [41]:
where \(\varepsilon_{H}\)and \(\varepsilon_{L}\) are the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energies, respectively.
Since the Bader theory of atoms in molecules (AIM) [42] has offered a valuable tool to understand the nature of a chemical bond in terms of electron density distribution, this theory is applied to analyze the electronic structure of the considered cluster. In the AIM theory, the critical points are classified as bond critical point (BCP), ring critical point (RCP) and cage critical point (CCP) according to their number and sign of their eigen values. The bond critical points are located along a maximum electron density trajectory that connects two atoms, known as bond path. The following parameters of electron density function are taken into account: electron density estimated at given CP (\(\rho\)), its Laplacian (\(\nabla^{2} \rho\)) being the sum of the three eigenvalues of the Hessian of electron density, the density of the total energy of electrons (H) and its two components, the kinetic (G) and potential (V) electron energy densities. The Laplacian of electron density is related to kinetic and potential electron energy densities as follow [43]:
It is well-known that small value of electron density at a given BCP (\({\rho_{BCP} }\) \(\le\)0.20) together with positive value of Laplacian of electron density at this point (\({\nabla^{ 2} \rho_{BCP} }\)>0) identified an ionic bond [44, 45].
3 Results and Discussions
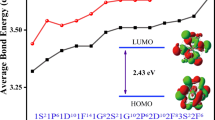
The optimized structure and DOS spectrum of pristine (BeO)12 nanocluster is presented in Fig. 1. This cluster includes six square and eight hexagon rings. As it is clear from Fig. 1 two kinds of individual Be–O bonds are distinguishable for the considered structure. One is shared between two hexagons (hh) with l hh = 1.53 Å and another between a square and a hexagon (hs) with l hs = l.58 Å. Note that the obtained vibrational frequencies for this cluster are in the ranges of 209–1,186 cm−1 which confirm the viability of this structure on the potential energy surface.

a The optimized geometry and b electronic density of states (DOS) spectrum of the pristine BeO nanocage
In this section the electronic properties of the (BeO)12 cluster are investigated. The obtained frontier molecular orbital energies (\(\varepsilon_{H}\)and \(\varepsilon_{L}\)) and the calculated HOMO–LUMO energy gap and hardness values for the considered (BeO)12 cluster are gathered in Table 1. The obtained energy gap for this cluster is about 7.55 eV implies to its insulator character. Also NBO calculations show that the natural charges of Be and O atoms are about 1.21 and −1.21 esu, respectively; hence an ionic character for this bond is expected. The validity of this claim is investigated through the atoms in molecules (AIM) theory.
The obtained critical points and bond paths of the pristine (BeO)12 nanocluster are depicted in the topological graphs (See Fig. 2). The calculated topological parameters at the obtained critical points are also summarized in Table 2. It is obvious from Fig. 2 that there are a CCP, two kinds of RCP as well as three kinds of BCP in the considered cluster. According to the Table 2 and the mentioned AIM criteria, Be–O bond critical points show ionic characters. Comparing electron densities at BCPs clarifies that hh-bond is stronger than hs-one (See Table 2).

The obtained molecular graphs for the pristine BeO nanocage. The yellow, green and small red balls indicate RCP, CCP and BCP respectively. The numbers in the critical points are listed in table 2
In this section the impact of carbon and silicon doping on the structural and electronic properties of (BeO)12 nanocluster is investigated. For this purpose, two individual sites (Be or O atom) for doping of the C and Si atoms are considered and a beryllium or oxygen atom of the considered cluster is substituted by carbon or silicon atom. It should be mentioned that CBe/SiBe denotes the (BeO)12 nanocluster in which one of the Be atoms of the cluster is replaced by one C/Si atom. Also, the notation CO/SiO refers to the structures in which one O atom is substituted by one C/Si atom. The optimized geometries of the considered doped-clusters are depicted in Fig. 3. All the considered clusters belong to C1 point groups. It should be noted that the structure of the doped-clusters changes dramatically due to the carbon or silicon doping; especially for the SiO doped cluster in which the cluster is opened due to the doping. Therefore carbon or silicon doping induces local deformation in the cluster and the doped atom protrudes out of plane in order to relieve tortional strain. This causes the elongation of the hs-bond length at the doping site from l Be-O = 1.58 Å to l C-O = 1.62 Å, l Be-C = 1.77 Å and l Si-O = 1.87 Å for CBe, CO and SiBe doped-clusters, respectively. The hh-bond length changes from l Be-O = 1.53 Å to l C-O = 1.43 Å, l Be-C = 1.73 Å and l Si-O = 1.77 Å. Also significant distortion in the bond angles are observed in Fig. 3. For instance the O–Be-O angle is changed from 97.82° to 93.81° and 99.06° in CBe and SiBe doped-clusters. Moreover the doping energy (\(E_{dop}\)) needed to introduce the C or Si atoms doping is calculated according to the following equation

The optimized geometries of (I) CBe, (II) CO (III) SiBe and (IV) SiO doped-(BeO)12 nanoclusters
where \(E_{[doped - cluster]}\) and \(E_{[pristine - cluster]}\) denotes the total energy of the pristine and doped cluster. \(E_{[Be//O]}\) is total energy for an isolated Be or O as well as \(E_{[C/Si]}\) presents total energy for an isolated C or Si. The obtained \(E_{dop}\)values are listed in Table 1. The results indicate that the doping process silicon doping needs more energy than carbon doping. However the doping energy for the CO cluster is -0.018 eV which shows that the formation of CO doped cluster is more favorable among all of these studied clusters.
The hybridization of Be, O, Si, C in the pristine and doped clusters are summarized in Table 3. According to this table, in the case of pristine (BeO)12 nanocluster, the NBO results show also that the hh-Be–O bond is formed by the interaction between a sp2.61 orbital on the beryllium and a sp1.70 orbital on the oxygen atom; while for the hs-Be–O bond consist of the interaction of a sp2.23 orbital on the beryllium and a sp2.21 orbital on the oxygen atom. Hence although the participation of orbital s and p on both Be atoms are very similar, the contribution of p orbital was increased for hs-Be–O bond than hh-Be–O bond. This confirms the observation that the bond lengths hs-Be–O (1.58 Å) is greater than hh-Be–O bond (1.53 Å).The obtained WBI are also in accordance with the obtained results (See Table 3). Also the natural ionicity index (\(i_{AB}\)) values for these bonds imply to their relative ionic nature. It is noticeable that for CBe, SiBe and CO doped clusters the hs-bonds of the doping site (C8-O15, Si8-O15 and C15-Be10, respectively) have less WBI and more bond length rather than hh-bonds (C8-O22, Si8-O22 and C15-Be10, respectively). This is mainly due to more p character of O15 and C15 atoms in these considered clusters (See Table 3). Furthermore, the obtained \(i_{AB}\) for the Be–O bonds near the doping site indicate that the ionicity of these bonds is approximately remained constant for these doped clusters. However in the case of SiO doped clusters the ionicity of Be8-O21 and Be2-O21 are slightly increased. The bond length/WBI of these considered bonds are also increased/decreased. This may due to this fact that the O21 atom used p orbital (90.54 %) in their bonds with Be. In the case of CBe, SiBe and CO doped clusters the elongation of Be–O bonds near the doping site is also related to more p character of oxygen and carbon atoms.
In order to investigate the variation of electron distribution during doping process, AIM analysis is performed for the considered doped-clusters. The obtained molecular graphs are depicted in Fig. 4. Furthermore the obtained topological parameters at the critical points are listed in Table 4. According to the this table, although electron density at the cage critical point (CCP) increases from 0.0014 a.u for pristine (BeO)12 cluster to 0.0016 a.u for CBe-doped cluster, it decreases to 0.0013 a.u for CO-doped cluster and it remains unchanged for SiBe and SiO doped clusters. Then silicon doping does not change the electron density at CCP; while replacing Be/O with a carbon atom increase/decrease the electron density at CCP. It has been also found that C or Si doping leads one RCP in square ring in which the electron density changes in the order CBe (\(\rho_{RCP}\)=0.0548 a.u) > CO (\(\rho_{RCP}\)=0.0497 a.u) > SiBe (\(\rho_{RCP}\)=0.0485 a.u) > SiO (\(\rho_{RCP}\)=0.0376 a.u). Hence beryllium substituting with carbon atom induces a considerable electron accumulation at square ring. Moreover the results of Table 4 show that the electron density at RCP of the hexagon ring beside the doping atom increases from 0.0155 a.u for pristine (BeO)12 -cluster to 0.0170 a.u for CBe-doped cluster; while it decreases to 0.0151, 0.0141 and 0.150 a.u for CO, SiBe and SiO doped clusters, respectively. Furthermore according to the mentioned AIM criteria it is obvious from Table 4 that the C–O bond in the CBe-doped cluster is covalent in nature (\({\nabla^{ 2} \rho_{BCP} }\)<0). However the considerable electron density at BCP of hh-C–O bond (\(\rho_{BCP}\)=0.2418 a.u) implies to its strong covalent character. Moreover the \({\nabla^{ 2} \rho_{BCP} }\)>0 and small values of electron density at BCP of C–Be and Si–Be bonds in CO- and SiBe-doped clusters indicate their ionic characters (See Table 4).

The obtained molecular graphs for (I) CBe, (II) CO (III) SiBe and (IV) SiO doped-(BeO)12 nanoclusters. The yellow, green and small red balls indicate RCP, CCP and BCP respectively. The labels of the critical points are listed in table 4
The molecular electrostatic potential (MEP) surface is an important descriptor in interpreting reactivity of sites for electrophilic and nucleophilic attacks. The MEP plot is the depiction of electrostatic potential mapped onto the total electron density surface. Since MEP is related to the total charge distribution of the molecule, it provides correlation between molecular structure with its physicochemical properties such as partial charges, dipole moment, and chemical reactivity. The obtained MEP surfaces for the studied clusters are depicted in Fig. 5. Red and blue areas in the MEP map refer to the regions of negative and positive potentials and correspond to the electron-rich and electron-poor regions, respectively; whereas the green color signifies the neutral electrostatic potential. The negative potential region of MEP plot is related to the region of electrophilic attack and positive potential region are related to the nucleophilic attack. According to the Fig. 5 a uniform distribution is observed for pristine (BeO)12. In all of the considered clusters, the Be atoms hold significant positive charges (electron deficient regions) and are favorable for the nucleophilic attack. Among all of the studied clusters, a maximum negative charge region (red color) is concentrated over carbon atom in the case of the CBe cluster and approximately over the silicon atom (yellow color) in the SiBe doped clusters. Therefore it seems that the reactivity of the (BeO)12 is increased toward of electrophilic attack by replacing Be atom with carbon and silicon atoms. However in the case of CO and SiO doped clusters no considerable change is observed in their MEP surfaces. Besides, the pictorial representation of the HOMO–LUMO distribution of the considered clusters is represented in Fig 6. The HOMO orbital primarily acts as electron donor, and LUMO orbital largely acts as electron acceptor. The interpretation from the HOMO–LUMO analysis in this figure is that in the case of pristine (BeO)12 cluster, the HOMO is concentrated over the oxygen atoms of the cage as well as the LUMO is spread over the beryllium atoms. Furthermore the HOMO is over the carbon and silicon atoms in the CBe and SiBe and C doped clusters, respectively; whereas the LUMO is mainly spread over the entire molecule except the carbon and silicon atoms. It is noticeable that according to the Fig 6, it seems that there is the tendency to the formation of triangles in these doped-nanoclusters. The wiberg bond order of the Be–Be (See Fig 6.) which caused the formation of triangles in the nanoclusters are checked. The calculated bond order values for these bonds are nearly less than 0.03. It is noticeable that no hybridization for Be–Be bond in clusters has been reported, confirming that they had electrostatic interactions and ionic nature.

The computed molecular electrostatic potential (MEP) for the (I) CBe, (II) CO (III) SiBe and (IV) SiO doped-(BeO)12 nanoclusters

Typically contour plots of HOMO of the of (I) pristine, (II) CBe, (III) CO (IV) SiBe and (V) SiO doped-(BeO)12 nanoclusters
In this section the electronic properties of the considered nanoclusters are analyzed from their density of states (DOSs) spectra. The obtained DOS spectra are shown in Fig. 7. This figure shows considerable decrease in energy gap due to the doping process. The energy gap of pristine (BeO)12 cluster is 7.55 eV which implies to its insulator character; but this energy gap reduces to 4.403, 2.301, 4.948 and 2.712 eV for the CBe, CO, SiBe and SiO doped-clusters, respectively. In other words the energy gap values decrease by 41.7, 69.5, 34.5 and 64.1 % for the CBe, CO, SiBe and SiO clusters upon doping process. It is obvious that the most considerable decrease is observed in CO-doped-(BeO)12 -nanocluster. According to Table 1 and the DOS plot in Fig. 7, the HOMO energy level changes in the range of 2.5-3.0 eV due to the doping process. Although negligible changes in the LUMO energies level are observed for CBe and SiBe doped(BeO)12 -nanocluster, the variation of Eg of CO-doped cluster is mainly due to the change of LUMO energy level (εL = -4.049). The changes in the energy gap due to the doping process, could be attributed to the sensitivity of the (BeO)12 nanocluster for a particular dopant and the electronic character of the cluster could be adjusted by specific impurity. In other words the energy gap of the (BeO)12 nanocluster could be engineered by introducing different dopants in the (BeO)12 cluster.

The obtained DOS spectra of the a CBe, b CO, c SiBe and d SiO nanoclusters
4 Conclusion
Density functional theory calculations are applied to investigate the structural and electronic properties of the (BeO)12 nanoclusters. The influence of carbon or silicon atom doping is also studied. It is shown that doping process induces local deformation at bond lengths which are scrutinized through NBO analysis. Also the electronic charge distributions are analyzed using Atoms in Molecules theory. The obtained DOS spectra indicate that C and Si doping decrease the energy gaps of the (BeO)12 nanocluster in order of, CO > SiO > CBe > SiBe by 69.5, 64.1, 41.7, and 34.5 % upon doping process. It seems that the electronic character of the (BeO)12 nanocluster could be adjusted by particular impurity.
References
S. Iijima, Nature 354, 56 (1991)
M.R. Provorse, C.M. Aikens, Comp. Theor. Chem. 987, 16 (2012)
M.L. Contreras, D. Avila, J. Alvarez, R. Rozas, Struct. Chem. 21, 573 (2010)
D. Hossain, C.U. Pittman, S.R. Gwaltney, J. Inorg. Organomet. Polym Mater. (2013). doi:10.1007/s10904-013-9995-6
M.T. Baei, J. Cluster Sci. 24, 749 (2013)
Z. Peralta-Inga, S. Boyd, J. Murray, C. O’Connor, P. Politzer, Struct. Chem. 14, 431 (2003)
A. Jain, V. Kumar, Y. Kawazoe, Comp. Mater. Sci. 36, 258 (2006)
L. Hong, H. Wang, J. Cheng, L. Tang, J. Zhao, Comp. Theor. Chem. 980, 62 (2012)
P.B. Sorokin, A.S. Fedorov, L.A. Chernozatonskii, Phys. Solid State 48, 398 (2006)
B. Baumeier, P. Kruger, J. Pollmann, Phys. Rev. B 76, 085407 (2007)
M. Zhao, Y. Xia, F. Li, R.Q. Zhang, S.T. Lee, Phys. Rev. B 71, 085312 (2005)
L.C. Ma, H.S. Zhao, W.J. Yan, J. Magn. Magn. Mater. 330, 174 (2013)
S. Duman, A. Sütlü, S. Bağcl, H.M. Tütüncü, G.P. Sirvastava, J. Appl. Phys. 105, 033719 (2009)
P. Pochet, L. Genovese, S. De, S. Goedecker, D. Caliste, S.A. Ghasemi, K. Bao, T. Deutsch, Phys. Rev B 83, 081403 (2011)
D.E. Bean, J.T. Muya, P.W. Fowler, M.T. Nguyen, A. Ceulemans, Phys. Chem. Chem. Phys. 13, 20855 (2011)
H.J. Xiang, Z.Y. Chen, J. Yang, J. Comput. Theor. Nanosci. 3, 838 (2006)
J.T. Muya, E. Lijnen, M.T. Nguyen, A. Ceulemans, Chem. Phys. Chem. 14, 346 (2013)
S. Polad, M. Ozay, Phys. Chem. Chem. Phys. 15, 19819 (2013)
R. Fabiański, L. Firlej, A. Zahab, B. Kuchta, Solid State Sci. 4, 1009 (2002)
R. Peköz, S. Erkoç, Comp. Mater. Sci. 46, 849 (2009)
A.F. Jalbout, Comp. Mater. Sci. 44, 1065 (2009)
J.H. Lee, B.S. Lee, F.T.K. Au, J. Zhang, Y. Zeng, Comp. Mater. Sci. 56, 131 (2012)
L. Ren, L. Cheng, Y. Feng, X. Wang, J. Chem. Phys. 137, 014309 (2012)
J. Beheshtian, Z. Bagheri, M. Kamfiroozi, A. Ahmadi, J. Mol. Model. 18, 2653 (2012)
J. Kakemam, A.A. Peyghan, Comput. Mater. Sci. 79, 352 (2013)
M.T. Baei, M.B. Tabar, S. Hashemian, Adsorpt. Sci. Technol. 31, 469 (2013)
I.R. Shein, M.V. Ryzhkov, M.A. Gorbunova, YuN Makurin, A.L. Ivanovskii, J. Exp. Theor. Phys. Lett. 85, 246 (2007)
M.A. Gorbunova, I.R. Shein, YuN Makurin, V.V. Ivanovskaya, V.S. Kijko, A.L. Ivanovskii, Physica E 41, 164 (2008)
A.D. Becke, J. Chem. Phys. 98, 5648 (1993)
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37, 785 (1988)
R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, J. Chem. Phys. 72, 5639 (1980)
J.P. Blaudeau, M.P. McGrath, L.A. Curtiss, L. Radom, J. Chem. Phys. 107, 5016 (1997)
U. Salzner, J.B. Lagowski, P.G. Pickup, R.A. Poirier, J. Comput. Chem. 18, 1943 (1997)
D.E. Wheeler, J.H. Rodriguez, J.K. McCusker, J. Phys. Chem. A 103, 4101 (1999)
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,V.G. Zakrzewski, J.A. Montgomery, Jr. R.E. Stratmann, J.C. Burant, S. Dapprich,J.M. Millam, A.D. Daniels, K.N. Kudin, M.C. Strain, O. Farkas, J. Tomasi, V.Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J.Ochterski, G.A. Petersson, P.Y. Ayala, Q. Cui, K. Morokuma, D.K. Malick, A.D.Rabuck, K. Raghavachari, J.B. Foresman, J. Cioslowski, J.V. Ortiz, A.G. Baboul,B.B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R.L.Martin, D.J. Fox, T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, C.Gonzalez, M. Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, J.L.Andres, C. Gonzalez, M. Head-Gordon, E.S. Replogle, J.A. Pople, Gaussian 03 Inc., Pittsburgh
F. Biegler-König, J. Schönbohm, D. Bayles, J. Comput. Chem. 22, 545 (2001)
N. O’Boyle, A. Tenderholt, K. Langner, J. Comput. Chem. 29, 839 (2008)
F. Weinhold, C. R. Landis, Discovering Chemistry With Natural Bond Orbitals, John Wiley & Sons, Hoboken (2012)
K.K. Pandey, G. Frenking, Eur. J. Inorg. Chem. 2004, 4388–4395 (2004)
J.F. Janak, Phys. Rev. B 18, 7165 (1978)
R.G. Parr, R.G. Pearson, J. Am. Chem. Soc. 105, 7512 (1983)
R.F.W. Bader, Atoms in Molecules: A Quantum Theory (Oxford University Press, Oxford, 1990)
C.F. Matta, R.J. Boyd, The Quantum Theory of Atoms in Molecules (Wiley, Weinheim, 2007)
V.G. Tsirelson, N.P. Tarasova, M.F. Bobrov, YuV Smetannikov, Heteroat. Chem. 17, 572 (2006)
J.T. Muya, E. Lijnen, M.T. Nguyen, A. Ceulemans, J. Phys. Chem. A 115, 2268 (2011)
Acknowledgments
The financial support of this work by Shahid Chamran University of Ahvaz is greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Shakerzadeh, E. A Theoretical Study on the Influence of Carbon and Silicon Doping on the Structural and Electronic Properties of (BeO)12 Nanocluster. J Inorg Organomet Polym 24, 694–705 (2014). https://doi.org/10.1007/s10904-014-0035-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-014-0035-y