
Abstract
Aggregation generally quenches the light emissions of chromophoric molecules. In this review, we demonstrate that 1,1-disubstituted 2,3,4,5-tetraphenyl siloles and 2,5-difunctionalized siloles as well as their polymers exhibit the opposite behaviors. Instead of quenching, aggregation has greatly boosted their photoluminescence quantum yields by up to two orders of magnitude, turning them from faint fluorophores into strong emitters. Such “abnormal” phenomenon of “aggregation-induced emission (AIE)” is attributed to restricted intramolecular rotations of the peripheral phenyl rings against the central silole core, which block the nonradiative channel via the rotational energy relaxation processes and effectively populate the radiative decay of the excitons. Utilizing such a novel effect, siloles and their polymers find an array of applications as: sensors for chemicals, explosives, pH, and biomacromolecules (proteins, DNAs and RNAs), indicators for determining CMC and monitoring layer-by-layer self-assembling, biocompatible fluorogens for cell imaging, visualizing agent for DNA gel electrophoresis, biolabels for immunoassay, stimuli-responsive organic nanomaterials, magnetic fluorescent nanoparticles for potential bio-imaging and -separation, and outstanding materials for efficient OLEDs and PV cells.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Siloles or silacyclopentadienes are a group of silicon-containing five-membered cyclic dienes. Since the first synthesis of a propeller-shaped silole molecule, 1,1,2,3,4,5-hexaphenylsilole (5, HPS, Scheme 1), by Braye and Hübel in 1959 [1, 2], exploration of the structures and functionalities in these fancy organometallic siloles has been an appealing topic among scientists from different research fields. Great efforts have been devoted to studies on the synthesis, reactivities, optoelectronic properties, coordination abilities of such compounds to transition metals, and the aromaticity of their anionic or cationic species [3, 4]. Compared with other common five-membered heterocyclic compounds such as pyrrole, furan, and thiophene, silole has the lowest LUMO energy level associated with the σ*–π* conjugation arising from the interaction between the σ* orbital of two exocyclic σ-bonds on the silicon atom and the π* orbital of the butadiene moiety [5–10]. Due to this excellent inherent attribute, siloles exhibit high electron acceptability and fast electron mobility and have been utilized as electron-transporting and light-emitting layers in the fabrication of electroluminescence (EL) devices [7, 11–15].

Synthesis of 1,1-disubstituted 2,3,4,5-tetraphenylsiloles
It is known that photoluminescence (PL) process is affected by both internal and external factors [16]. For example, internally or structurally, a molecule with a rigid conformation can emit more efficiently than its flexible congener, because active intramolecular motions, such as rotations and vibrations, can effectively annihilate the excited states nonradiatively. Externally or morphologically, aggregation of luminophoric molecules can quench their PL, due to the increase in the short-range molecular interactions and the formation of detrimental species, such as excimers and exciplexes [17–23]. On the other hand, aggregation can positively restrict the intramolecular motions of the luminophores, which block the nonradiative decay channels and enhance their PL efficiencies. Whether aggregation quenches or boosts a PL process depends on which of the two antagonistic effects prevails in a particular system. Unfortunately, the negative effect of aggregation dominates the PL processes of most luminophoric molecules. Thus, it has become a “common knowledge” known as aggregation-caused quenching (ACQ) of light emission. Luminophore aggregation is an inherent process accompanying film formation. The ACQ effect therefore have been a thorny obstruct to the development of efficient organic light-emitting diodes (OLEDs), in which the luminophoric materials are used as the emitting layers in the film state.
In 2001, our group discovered such a system, in which luminophore aggregation played a constructive, instead of destructive, role in the light-emitting process: a series of silole molecules were found nonluminescent in the solution state but emissive in the aggregate state (as nanoparticle suspensions in poor solvents or as thin films in the solid state) [24]. The AIE effect greatly boosts their PL quantum yields (ΦF’s) by up to two orders of magnitude, turning them from faint into strong emitters [24–26]. We coined “aggregation-induced emission” (AIE) for this novel phenomenon, because the nonluminescent silole molecules were induced to emit by aggregate formation. The novel AIE effect is exactly opposite to the notorious ACQ effect discussed above. The phenomenon is of academic value: whilst there are matured theories to explain the ACQ effect, new models will need to be established to understand the “abnormal” AIE effect. The phenomenon is also of practical implications: in the AIE system, one can buoyantly enjoy the advantages of aggregate formation, rather than work against it. It will permit the use of dye solutions with any concentrations for bioassays and enables the development of “turn on” or “light up” nanosensors and photoelectronic devices by taking advantage of luminogenic aggregation [25–44]. Indeed, by utilizing this AIE property, we have fabricated silole-based OLEDs, which exhibit outstanding EL performances. A diode of HPS, for example, radiates brilliantly (luminance up to ~60,000 cd/m2) [26], while that of 1-methyl-1,2,3,4,5-pentaphenylsilole (4, MPPS, Scheme 1) shows an extremely high external quantum efficiency (ηEL = 8%) [25], reaching the theoretical limit for a light-emitting diode based on a singlet emitter.
Attracted by these fascinating perspectives, we have launched a program directed towards the decipherment of the working principle behind the AIE process of siloles. In this review, we summarize our research work on the development of siloles (Schemes 1, 2, 3, 4, 5, 6) and silole-containing linear (Chart 1) and hyperbranched polymers (Schemes 7, 8). We tell why they are AIE-active and their practical applications.

Synthesis of x,y-bis(2,6-diisopropyl)phenyltetraphenylsiloles (HPS-iPrx,y)

Synthesis of 1,1-disubstituted 2,3,4,5-tetraphenylsiloles by post-functionalizations

Synthesis of 2,5-disilylsiloles

Synthesis of 2,5-diarylsiloles

Synthesis of 2,5-diethynylsiloles

Synthesis of hyperbranched poly(1,1-silolylphenylene)s by diyne/monoyne copolycyclotrimerization

Synthesis of hyperbranched poly(2,5-silolylphenylene)s by diyne homopolycyclotrimerization

Substituted polyacetylenes bearing silole pendants
2 Synthesis
The 1,1-difunctionalized 2,3,4,5-tetraphenylsiloles (Schemes 1, 2, 3) [26, 42, 44] as well as 2,5-difunctionalized siloles (Schemes 4, 5, 6) [7] are synthesized according to published procedures. The 1,1-disubstituted siloles are prepared by lithiation of diphenyacetylene, followed by reactions of the intermediate dilithiotetraphenylbutadiene with appropriate silane chlorides. The commercial availability of many symmetric (X = Y) and asymmetric (X ≠ Y) dichlorosilanes (Cl2SiXY) greatly facilitates our preparations of siloles with different combinations of X and Y groups at the 1,1-positions. Alternatively, reactions of dilithio-1,2,3,4-tetraphenylbutadiene with Cl3SiX and Cl4Si will yield chlorosiloles with one and two chloro groups at the 1- and 1,1-positions, respectively. The chloro groups can be easily replaced by nucleophiles (X and Y) to provide various functional 1,1-disubstituted siloles. By means of post-reactions, 1-ethynylpentaphenylsilole (6, EPPS) can be highly functionalized to furnish 23–27 with amino (23, Am-EPPS), hydroxy (24, HO-EPPS), ferrocenyl (25, Fc-EPPS), carbazolyl (26, Cz-EPPS) and anthracenyl (27, An-EPPS) groups by Sonogashira coupling, respectively (Scheme 3) [42]. Miscellaneous 2,5-difunctionalized siloles can be prepared from diethynylsilane precursors. They undergo intramolecular reductive cyclization in an endo–endo mode upon treatment with lithium naphthalenide (LiNaph) to form 2,5-dilithiosiloles, which convert into the corresponding 2,5-disilylsiloles, 2,5-diarylsiloles, and 2,5-diethynylsiloles in high yields, respectively, by (i) trapping with electrophiles, e.g., various chlorosilanes (Scheme 4) [45], (ii) transmetalation with ZnCl2 · TMEDA (TMEDA = N,N,N′,N′-tetramethylethylenediamine) to give 2,5-dizinc siloles followed by subsequent reaction with appropriate aryl bromides in the presence of Pd(0) catalyst (Scheme 5) [7], and (iii) treatment with NBS to yield 2,5-dibromosiloles followed by Pd(0)-catalyzed cross-coupling reaction with appropriate alkynes (Scheme 6) [46]. The substituents at different positions of the siloles can be further modified. For example, tertiary amine at 2,5-positions of 57 can be ionized in the presence of ethyl bromide to offer quaternary ammonium salt 58. Theoretical calculations show that the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) of tetraphenyl- and hexaphenylsiloles are mainly localized on the silole ring and two aromatic groups at 2,5-positions, while the LUMOs have a significant orbital density at the two exocyclic Si–C bonds [36]. Indeed, the emission colors can be tuned from blue to red by changing the groups at 2,5-positions. For example, emission of blue (28–36), green (1–27, 37–42, 50–53, 57–60, and 63–70), yellow (54–56, 61, and 62), and red (43, 44, 47, and 48) have been achieved.
Our group has been working on acetylene chemistry for more than 10 years and is experienced in preparing polymers constructed from triple-bond building blocks [47–49]. We are interested in incorporating siloles into conjugated polymers, in an effort to create new materials that are processable with novel optoelectronic properties. Through elaborate efforts, we succeeded in synthesizing silole-containing polyacetylenes using metathesis polymerizations [27] (Chart 1) and hyperbranched polymers by alkyne homo- and cocyclotrimerizations [28, 48] (Schemes 7, 8). All the silole-containing polymers are completely soluble in common solvents and enjoy high thermal stability.
3 Phenomenon: Aggregation-induced Emission
Our research group has been interested in employing acetylenic triple-bond building blocks to construct new molecules and polymers with linear and hyperbranched structures and advanced functional properties. In our seeking for highly emissive linear and hyperbranched polymers, we were attracted by the “abnormal” light-emitting behavior of siloles [24]. We prepared HPS [26] according to the synthetic route shown in Scheme 1. Before purifying the reaction product HPS by silica-gel column, we run a thin-layer chromatography (TLC) test. When the TLC plate was taken out from the development tank, hardly could any spots be seen in the wet plate under illumination of a UV lamp. After the solvent was evaporated from the plate, a green light-emitting spot became visible. Thus, the fluorescence was off and on in the wet and dry plates, respectively, implying that the HPS molecules are nonemissive when dissolved but become luminescent when aggregated. This extraordinary observation spurred us to further investigate its luminescence behaviours.
HPS is very soluble in acetonitrile, tetrahydrofuran (THF), and chloroform, slightly soluble in methanol, but completely insoluble in water. Solutions of HPS in its good solvents are nonemissive, as revealed by the example shown in Fig. 1 for its solution in acetonitrile. Addition of a large amount of water into the acetonitrile solution of HPS causes the silole molecules to aggregate and induces them to emit efficiently. This duly substantiates the observation in the TLC test: HPS is induced to emit by aggregation. In other words, it is AIE active.

(Left) Chemical structure of HPS. (Right) HPS in the acetonitrile/water mixtures containing different volume fractions of water; photographs taken under UV illumination
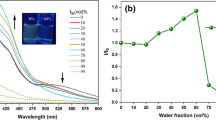
The intriguing AIE phenomenon was further confirmed by spectroscopic analyses. The PL spectrum of the HPS solution in acetonitrile is nearly a flat line parallel to the abscissa (Fig. 2a), with a ΦF as low as 0.22%. The ΦF value remains unchanged until ~50% water is added into the solution. Afterwards, the ΦF value increases swiftly (Fig. 2b). In the solvent mixture with 99% water, the ΦF value of HPS rises to ~56%, which is ~255-fold higher than that in the pure acetonitrile solution. The AIE effect has enabled HPS to emit efficiently in the solid state: the emission efficiency of a thin film of HPS (ΦF,f) is as high as 78%, as estimated by using a tris(8-hydroxyquinolinato)aluminum(III) (AlQ3) film as standard [36].

a PL spectra of HPS in acetonitrile/water mixtures with different water fractions. b Quantum yield (ΦF) of HPS versus solvent composition of acetonitrile/water mixture. ΦF for a thin film of HPS (ΦF,f) is given in panel b for comparison
Is the AIE effect an isolated phenomenon for HPS alone or a general property for the silole family? To answer this question, we prepared a variety of silole derivatives and examined their photophysical properties in both solution and aggregate states. All the derivatives behave like HPS. For example, MPPS was nonluminescent when dissolved in good solvents (e.g., ΦF = 0.06% in ethanol) but became highly emissive when aggregated in poor solvents or fabricated into thin film (ΦF,f = 85%) [36]. The aggregation has boosted its emission efficiency by as much as 1417 times!
The silole derivatives share the following common features of photophysical behaviours: (i) In the good solvents or their aqueous mixtures with “low” water contents (≤50%), they are nonemissive, with their ΦF values in the vicinity of 0.1%, (ii) in the solvent mixtures with “medium” water contents, their emissions are progressively intensified with increasing water contents, and (iii) in the solvent mixtures with “high” water contents (>80%), their PL spectra bathochromically shift.
In all the solvent mixtures, including those with high water contents, the silole derivatives give similar absorption spectra. Their spectral profiles experience little red shifts, as can be seen from the example shown in Fig. 3a for the HPS solutions. The spectra of HPS measured in the solvent mixtures with high water contents show absorption tails well extending into long wavelength region. This implies that the HPS molecules have aggregated into nanoparticles in the aqueous mixtures, because it is well known that the Mie effect of nanoparticles causes such level-off tails in the absorption spectra.

a Absorption spectra of HPS in acetonitrile/water mixtures with different water fractions. Size distributions of the nanoparticles of HPS in the acetonitrile/water mixtures containing b 80% and c 90% water
Particle size analyses reveal the existence of particles with average sizes of 190 and 130 nm in the solvent mixtures with 80% and 90% water, respectively (Fig. 3), confirming that the HPS molecules have indeed aggregated into nanoparticles. In the solvent mixtures with water contents ≤50%, no level-off spectral tails in the long wavelength region are recorded by the UV–vis spectrometer and no nanoparticles are detected by the particle size analyzer, proving that the HPS molecules are genuinely dissolved as isolated species in the solvent mixtures with low water contents.
4 Mechanism
What is the real cause or mechanism for the “abnormal” AIE phenomenon? Since the discovery of this novel effect by our group in 2001 [24], we have devoted great efforts to deciphering its working principle, which may help gain new photophysical insights and spawn technology innovations. We considered a number of possible mechanistic pathways, including conformational planarization, J-aggregate formation, and twisted intramolecular charge transfer (TICT), none of which, however, is supported by the experimental data.
If the conformation of HPS molecules became more planar upon aggregate formation, both of absorption and emission spectra would bathochromically shift. The spectra of HPS, however, do not change much in the mixtures with different water contents (up to ~80%; Figs. 2, 3). Crystal structure analysis reveals that the conformation of the HPS molecules maintain twisted in the solid state. These experimental observations exclude the possibility that the conformational planarization is a major cause of the AIE phenomenon.
The silole derivatives are nonpolar molecules. They contain no polar moieties such as donor (D) and acceptor (A) groups and hence experience no D–A or push–pull interactions. This structural feature makes it unlikely for the silole molecules to form J-aggregates, as verified by the X-ray diffraction (XRD) measurements.
TICT [50] has been known to greatly affect the emission properties of some chromophoric molecules containing D and A groups. The prime example of great interest is 4-N,N-dimethylaminobenzonitrile (DMABN) (Scheme 9). In the ground state, the molecule is almost planar, which corresponds to the maximum conjugation between the dimethylamino group and the phenyl ring. According to the Franck–Condon principle, the molecule in the locally excited state (LE) is still planar, but solvent relaxation takes place with a concomitant rotation of the dimethylamino group until it is twisted at a right angle and the conjugation is lost. In the resulting TICT state, which is stabilized by the polar solvent molecules, there is a total charge separation between the dimethylamino group and the cyanophenyl moiety. In addition to a fluorescence band due to the emission from the LE state, a band corresponding to the emission from the TICT state is observed at longer wavelengths. In such a system, polarity of the solvent has been found to alter both the ground and excited states of the molecules. Increasing the polarity of solvents of DMABN solutions will red-shift its peak absorption (λab) and emission (λem). Especially, its λem is more sensitive to the solvent polarity. Normally, in nonpolar solvent, there is only one emission peak from the LE state, while two emission peaks appear in polar solvent from the LE and TICT states, respectively, in which the latter peak is red-shifted by up to >100 nm from the former one. TICT state is, in most cases, more likely to relax by nonradiative processes, thus leading to the lower PL intensity and ΦF value of DMABN in polar environments. Thus, the emission properties of this group of molecules highly depend on the solvent polarity.

Transition of DMABN between locally excited (LE) state and twisted intramolecular charge transfer (TICT) state through intramolecular rotation
However, the solvent polarity has little effect on the absorption and emission of siloles. Table 1 shows the UV absorption of HPS in different solvents [26]. The solvent polarity, even when varied by ~3.7 times on the Debye scale, exerts little effect on their absorption transitions, as exemplified by the very small changes in the λab of the silole solutions. We measured the PL spectra of the siloles in a variety of solvents with very different polarities (cyclohexane, toluene, dichloromethane, chloroform, carbon tetrachloride, ethyl acetate, THF, dioxane, acetone, acetonitrile, methanol, ethanol, etc.). Similarly, changing the solvent polarity hardly causes any changes in their emission transitions. All the solutions almost give the same spectral profile, irrespective of the polarity of the solvents. These results evidently rule out the likelihood of the involvement of the TICT mechanism in the AIE process of the silole system.
Fundamental physics teaches us that any molecular motions consume energy. As can be seen from the left panel of Fig. 1, six phenyl peripheries are attached to one silole core in HPS; the former (rotors) can rotate against the latter (stator) via the single-bond axes. In the solutions, the intramolecular rotation is active, which serves as a relaxation channel for the excited state to decay. In the aggregates, the rotation is restricted due to the physical constraint, which blocks the nonradiative path and activates the radiative decay. We thus hypothesize that the restriction of intramolecular rotation (RIR) is a cause for the AIE effect (Chart 1) [26]. To check the validity of this hypothesis, we performed a series of tests to externally and internally modulate the RIR process.
4.1 Viscochromism
As an external control, we first checked the effect of solvent viscosity on the silole emission [26]. The RIR process is expected to be affected by the medium viscosity: the more viscous the medium, the slower the intramolecular rotation and hence the stronger the silole emission. Glycerol is a very viscous liquid, whose viscosity at 25 °C is about 1,720-fold higher than that of methanol. Blending glycerol with methanol at different ratios therefore can afford solvent mixtures with drastically different viscosities.
As expected, the fluorescence of HPS is intensified with an increase in the viscosity of the solvent mixture (Fig. 4). In the mixtures with glycerol fractions of <50%, the PL intensity increases “linearly” with viscosity on the semilog scale. The emission enhancement in this region should be predominantly due to the viscosity effect (viscochromism), because the silole molecules are soluble in these mixtures (cf., Fig. 2b). In the mixtures with glycerol fractions of >50%, the PL intensity mounts rapidly, due to the aggregate formation caused by the poor solubility of the silole molecules in the mixtures with high glycerol contents.

PL peak intensity of HPS versus composition of glycerol/methanol mixtures; [HPS] = 10 μM
4.2 Thermochromism
Since cooling can also fortify RIR process, we investigated temperature effect on the silole emission [26]. THF is used in the study because of its strong solvating power and low melting point (−108 °C), which can help keep the solute molecules in the solution state at low temperatures. When a THF solution of HPS is cooled, its emission intensity is increased (Fig. 5a). The spectral profile and peak position are virtually unaffected by lowering the temperature, indicating that the emission is still associated with the radiative decay of the singlet excitons but not the triplet ones.

a PL spectra of HPS in THF at different temperatures; [HPS] = 10 μM. b NMR peaks of phenyl protons of HPS in dichloromethane-d 2 at different temperatures
THF has a low viscosity (0.456 cP at 25 °C) with a small temperature coefficient (~0.008 cP/K). The enhancement in the PL intensity above the melting point of the solvent thus should not be due to viscosity but temperature effect. Cooling suppresses the thermally susceptible intramolecular rotations, thus intensifying the HPS emission. Cooling the THF solution by liquid nitrogen to −196 °C brings about a big jump in the PL intensity. The liquid solution must have been frozen into a solid “glass” at such a cryonic temperature. In the solid matrix, molecular rotations are greatly restrained and the PL is hence dramatically enhanced.
To experimentally verify the RIR process, dynamic NMR spectra are taken. The fast conformational exchange caused by the rapid intramolecular rotation at room temperature leads to sharp NMR peaks, which can be broadened by cooling, as the rotation slow down at lower temperatures. The HPS solution in dichloromethane exhibits sharp NMR resonance peaks at ambient temperature, which are broadened with a decrease in temperature (Fig. 5b). The dynamic NMR spectral data thus confirm that the intramolecular rotations have indeed been restricted at the low temperatures.
Following the same principle, it is envisioned that heating the siloles in the solid state would weaken the emission. We carried out a simple heating-cooling cycle to monitor the transition of the emission intensity of solid HPS under UV illumination. Figure 6 reveals that the solid powder of HPS emits strongly at room temperature, while the melt is nonemissive. After cooling it down, its strong emission is recovered. This can be explained that when the siloles are in the melt state, the vigorous intramolecular motions (rotation, vibration, and so on) deactivate the excitons efficiently, hence leading to no emission.

Switching the emission of HPS by a heating–cooling cycle. Photographs taken under UV illumination at a room temperature (original solid powder), b ~200°C (melt liquid), and c room temperature
4.3 Piezochromism
The pressurization exerts complex effects on the HPS luminescence [43]. The emission is first enhanced to a plateau in a very fast speed upon pressurization, showing a unique effect of pressurization-enhanced emission or piezochromism (Fig. 7). However, when the HPS film is further pressurized, its emission is slowly decreased from the plateau. Theoretically, pressurization imposes antagonistic effects on the light emission. Pressurization brings molecules to closer neighborhood. The reduced distance between the dye molecules decreases the freedom of their molecular motions and strengthens the RIR process. On the other hand, it boosts intermolecular interactions and promotes the excimer formation. The RIR process enhances, but the excimer formation weakens, light emission.

Effect of externally applied pressure on the PL intensity of HPS films. Data for an AlQ3 film is shown for comparison
The latter effect, i.e., the pressurization-caused quenching, has been commonly observed in the conventional luminophore systems. This is manifested by the result obtained from the control experiment on a solid film of AlQ3: its PL intensity is monotonously weakened with increasing pressure. This is probably due to the pressurization-enhanced π–π stacking interactions between the plate-like quinoline rings. It is worth noting that the intramolecular rotation in an AlQ3 molecule is prohibited because its quinoline rings are rigidly held together by multiple bonds between the aromatic (quinoline) rings and the metal (aluminum) center.
The benzene rings in an HPS molecule, however, can rotate because its phenyl peripheries are linked to the silole core via single bonds. The steric effect between the phenyl rings forces the molecules to take a twisted propeller-shaped conformation, which are difficult to assume a dense packing structure when they are deposited onto the substrate by the vapor deposition process. In the molecular voids generated by the loose packing, the HPS molecules can still undergo some limited rotational motions. Pressurization decreases the void volumes and densifies the molecular packing. This strengthens the RIR process and thus boosts the silole emission. Further pressurization, however, brings the silole molecules to such a close neighbourhood that excimer formation becomes possible. This may explain why the silole emission slowly drops from the plateau in the high pressure region.
The molecular motions of HPS mainly consist of rotation (e.g., CPh–Csilole torsion) and vibration (e.g., CPh–Csilole stretching and bending). The change in the solvent viscosity mentioned above can hardly affect the vibrational motion. The pressurization in the low pressure region can shorten the distance between, but exert little effect on the distance within, the silole molecules [51]. The piezochromism observed in the low pressure region should be mainly caused by the suppression of the molecular rotation rather than vibration.
4.4 Fluorescent Decay Dynamics
Lifetime is an important kinetic parameter for PL decay. The emission behaviours of HPS are studied using a time-resolved technique [30]. Addition of water into the HPS solution not only enhances the light emission but also leads to the dramatic change in the emission lifetime, as illustrated by the time-resolved fluorescence curves in Fig. 8. The lifetime data is summarized in Table 2. In pure DMF, the excited-state relaxes in a single-exponential way, with all the excited molecules decay through the same pathway. The PL lifetime is 40 ps, close to the resolution limit of the apparatus (~25 ps). With addition of water into DMF, the excited state starts to decay via two relaxation pathways. In the mixture with 30% water, 80% (A 1) and 20% (A 2) of the excited HPS molecules decay via the fast and slow channels with lifetimes of 0.10 ns (τ1) and 3.75 ns (τ2), respectively (Table 2, no. 2). With an increase in the water content in the solvent mixture, the decay through the fast channel is slowed down, while the excited molecules decaying through the slow channel is populated. In the mixture with 90% water, the excited states mainly decay via the slow channel and the lifetime for the slow component is as long as 7.16 ns (Table 2, no. 4).

Time-resolved fluorescence of HPS in DMF/water mixtures
We also carried out lifetime measurements of HPS in solvents with different viscosities (Table 2, nos. 5–8) and in solutions at different temperatures (Table 2, nos. 9–13). Their fluorescence decay behaviors agree well with their PL intensity changes in solution-thickening and -cooling experiments. In the less viscous solvent at room temperature, the silole molecules are dissolved as isolated species. Little restriction is imposed on their intramolecular rotations. The A 1 value is thus a unity, indicating that the HPS molecules are all in the same environment and decay through the same pathway. The decay is very fast due to the efficient decay process associated with the active intramolecular rotation. Increasing solvent viscosity and decreasing solution temperature strengthen the RIR process and activate the radiative decay channel. All these results strongly support the RIR model for the AIE effect.
The theoretical work carried out by Shuai’s group has further verified our RIR hypothesis [52, 53]. Employing the first-principle calculation to simulate the excited states, the researchers have found that the low-frequency twisting motions of the phenyl peripheral rings of siloles lead to nonradiative decays, which can be alleviated by solution thickening, aggregate formation, and solid-state packing.
4.5 Internal Structural Tuning
All the external control experiments described above prove that the AIE effect stems from the RIR process and that the silole emission can be modulated by physical and engineering manipulations. We proceeded further and tried to exercise internal control on the silole emission. It is rationalized that if our proposed RIR model is correct, impeding intramolecular rotations by chemical means or structural design at molecular level should make the silole luminophores emissive even in the solution state.
To molecularly activate the RIR process, we attached two isopropyl groups to the peripheral phenyl rings of HPS to offer a group of silole regioisomers HPS-iPx,y (22), in which HPS-iP2,4 and HPS-iP3,4 are the least and most spaciously crowded, respectively [32]. Different from its HPS parent, HPS-iPx,y is luminescent in solutions, although its PL efficiency varies dramatically with its regiostructure (Fig. 9). In acetone, the regioisomers of HPS-iPx,y emit blue-green lights, whose ΦF’s increase in the order of HPS-iP3,4 > HPS-iP2,5 > HPS-iP2,4. Similar results are obtained in such a solvent as THF, confirming that the ΦF order observed in acetone is of generality. The ΦF’s of HPS-iPx,y are higher than those of “normal” siloles with ΦF of ~0.1% being most typical. The rotation barriers (E R’s) experienced by the aryl rings in HPS-iPx,y are generally higher than that in HPS (Table 3). The E R’s of the iPr-substituted phenyl rings are especially high (E R,Ph′ ≥ 50 kcal/mol), practically inhibiting their intramolecular rotation processes [54–56]. It is known that a more rigid chromophore is often a more efficient emitter [56–62]. The structural rigidification caused by the high rotation barriers has clearly played a decisive role in making HPS-iPx,y more emissive than its HPS parent. Time-resolved experiments give well consistent results with the change order of ΦF. This internal control experiment evidently demonstrates that the PL behaviours of the silole molecules can be readily tuned by structural tailoring or molecular engineering.

a PL spectra of acetone solutions of HPS-iPx,y (22; 10 μM) and b–d photographs of the HPS-iPx,y solutions taken under UV illumination (365 nm)
4.6 AIE-active Silole-containing Polymers
Taking the considerations that siloles possess valuble AIE property and our group has strong background and experience in acetylene polymer chemistry, we incorporated silole moieties into the polyacetylenes as pendants with an expectation to obtain AIE-active polymers with good processability (cf., Chart 1) [27]. As anticipated, they are AIE-active. For example, the molecularly dissolved species of P19 (e.g., in chloroform) is practically nonemissive but its nanoaggregates in poor solvents (e.g., a 90% methanol/chloroform mixture) and solid films are highly luminescent (Fig. 10); that is, the emission is induced by the aggregate formation. Obviously, the AIE effect occurs in this system: the ΦF (~9.3%) of the nanoaggregate of P19 in a methanol content of 90% is ~46 times higher than that of its molecularly dissolved species.

PL spectra of P19 in chloroform (molecularly dissolved solution), a methanol/chloroform mixture (9:1 by volume; nanoaggregate suspension), and solid state (thin film)
Further, an aggregation-induced emission enhancement (AIEE) phenomenon is observed in hyperbranched poly(2,5-silole)s by polycyclotrimerization of silole diyne monomers (cf., Scheme 8) [48]. The diyne monomers are nonemissive in solutions, while their polymers are already luminescent when molecularly dissolved in organic solvents. An example of the PL spectrum of hb-P50 is given in Fig. 11. The silole units in the hyperbranched polymer are knitted by rigid benzene rings and located within the polymer skeleton, which restricts their intramolecular rotations of peripheral benzene rings around the central silole core and thus makes the polymer emissive in the solution state. It can be seen that hb-P50 has much stronger emission than its monomer 50 at the same concentration. Gradual addition of water into the THF solution of hb-P50 has progressively enhanced its light emission, demonstrating a novel AIEE effect. This example provides us a further evidence to understand the AIE phenomenon.

PL spectra of hb-P50 and its monomer 50 in THF/water mixtures with different fractions of water; [hb-P50] = [50] = 10 μM
4.7 Morphochromism
One issue is still outstanding: why does the PL spectrum of HPS red shift when the water fraction in the acetonitrile/water mixture is increased from 80% to 90% (cf., Fig. 2a)? Careful inspection of the absorption spectra of HPS shown in Fig. 3a reveals that the light-scattering effect in the long wavelength region is stronger in the solvent mixture with 80% water than that in the mixture with 90% water. The particle size analysis indicates that the aggregates in the former mixture are bigger in size (196 nm) than those in the latter (130 nm; cf., panels B and C of Fig. 3).
We speculate that the aggregates in the solvent mixtures have different morphology structures. In solvent mixtures with “low” water contents, the silole molecules may steadily assemble into crystalline clusters, whereas in the mixtures with “high” water fractions, the molecules may abruptly agglomerate into amorphous aggregates. Transmission electron microscope (TEM) and electron diffraction (ED) measurements confirm that this is indeed the case: the silole aggregates formed in the mixtures with “low” and “high” water contents are crystalline and amorphous, respectively [34].
Additionally, the aggregates of HPS-iP2,4 in the crystalline and amorphous films both show blue-shifted spectra, with their respective λmax’s at 461 nm and 481 nm (cf., Table 3). The solid films of HPS-iP2,5 and HPS-iP3,4 also give blue-shifted spectra in comparison to those of their dilute solutions, confirming that the aggregation-induced blue shift is a general property of all the siloles. In the solid aggregates, the intramolecular rotational processes of the siloles are more restricted and the aryl peripheries may be more twisted from the central silole cores, in comparison to those in the dilute solutions. The former effect (enhanced rigidity) increases the luminescence efficiency, while the latter effect (reduced conjugation) may blue-shift the emission spectra.
Crystallization usually bathochromically shifts PL spectra, but why is the opposite effect observed in the silole system? Analyses of the conformation structure and packing arrangement of the HPS molecules in the crystalline phase offer some clues. In the crystals, the silole molecules take a highly twisted fan-like conformation (Fig. 12a). The peripheral phenyl rings are twisted out of the silacyclopentadiene plane to varying extents, with typical dihedral angels of ~30o for the phenyl plates at the ring positions next to silicon atom (C2 and C5 positions) and larger twists of ~70o at the C3 and C4 positions. The sp 3 hybridization of the silicon atom makes the phenyl rings attached to it at the Si1 position completely out of plane of the silole core. The nonplanarity caused by the steric repulsion between the neighboring phenyl plates at the ring carbon atoms as well as the sp 3 hybridization of the silicon atom at the ring bridge reduces the intermolecular interactions and the likelihood of excimer formation. The physical confinement in the crystal lattice rigidifies the molecular conformation of HPS, thereby increasing its PL efficiency in the solid state.

a ORTEP drawing of HPS. b Packing diagrams of HPS crystals, where the interplane distance is 10.04 Å and the intermolecular distance within the unit cell is 7.61 Å
The unusual blue shift observed in the crystalline phase is attributable to the conformation twisting in the crystal packing process, during which the silole molecules conformationally adjust themselves in order to fit into the crystalline lattice. Without this restraint, the silole molecules in the amorphous phase may assume a more planar conformation and thus show a redder luminescence. The conformational difference in the amorphous and crystalline phases is evidently the origin of the unusual morphochromism. The large interplane (~10 Å) and intermolecular (~7.6 Å) distances in the crystal unit cell (Fig. 12b) manifest the lack of strong chromophore interactions that tend to induce nonradiative relaxations and red-shifts as seen in the conventional crystals with π–π stacking interactions.
In summary, all the designed experiments above provide evidences to verify the AIE hypothesis of the silole molecules. Normally, traditional dyes suffer from ACQ effect originating from the formation of excimer or exciplex in the aggregation state. However, aggregation effect, acting as a positive role, greatly enhances the light emission efficiency of siloles. What does aggregation do?
When a fluorophore is excited, without the occurrence of photo-induced reactions (e.g., solvolysis, cis–trans (E–Z) photoisomerization, photo-induced electron transfer, excited-state proton transfer and so on), radiationless transition of the excited state (or exciton) can be via internal conversion (IC) through singlet excited state to singlet ground state (S1–S0), or intersystem crossing through singlet excited state to triplet state (S1–T1) relaxation processes according to the Perrin–Jablonski diagram [50]. The S1–S0 and S1–T1 nonradiative relaxations are normally mediated by rotational and vibrational motions of the molecule. Thus, restriction of these motions will decrease the nonradiative recombination rate of the excited state and enhance the emission efficiency. For S1–T1 intersystem crossing, the energy transfer process is very sensitive to the energy level of S1 to T1. The probability for such event is increased if the extent of overlap of vibrational levels of these two states becomes higher. The spin-orbit coupling constant in silole 1 between S1 and T1 is calculated at CAS (4,4)/6-31G level within the quantum chemical package Gamess to be 1.6 cm−1, which is too small as compared with the radiative decay rate [36]. This implies that IC is the major way of the nonradiative decay in these silole molecules. We know that ΦF = τ/τ r , where PL lifetime τ is related to radiative (τ r ) and nonradiative (τ nr ) lifetimes by the expression 1/τ = 1/τ r + 1/τ nr (k = 1/τ; k = k r + k nr , where k r and k nr are radiative and nonradiative decay rate, respectively). The τ r is the intrinsic property of the molecule. Consequently, the higher the nonradiative decay rate is, the lower is the ΦF. In the solutions, the rotation of the peripheral phenyl rings against the central silole core deactivates the excited states via the rotational energy relaxation. The energy can be transferred to the solvent during collisions of the excited molecule with the surrounding solvent molecules. External (viscosity, temperature, and pressure) or internal (structural control) perturbation on the silole molecules restricts their rotations, which block the nonradiative decay channel and enhances their light emission efficiency (Scheme 10). From the crystal structures of siloles (Figs. 12, 13) [38], we can see that the aggregates of these propeller-shaped siloles are not normal H-, J-, or X-aggregates, but special aggregates with large intermolecular distances, which prevent them from suffering the emission quenching due to the π–π stacking in the solid state. The molecular conformations of the silole crystals are locked and stabilized by multiple C–H···π hydrogen bonds, which hamper the aromatic rings from undergoing conformational changes caused by such movements as intramolecular rotations. This structural rigidification has made the silole crystals strong emitters.

Schematic illustration of aggregation-induced emission (AIE) mechanism

Perspective view of the packing arrangements in crystals of siloles (a) MPPS (4), (b) 9, (c and d) 6. The aromatic C–H···π hydrogen bonds are denoted by dotted lines
Although TICT and our proposed AIE mechanism are both related to rotational relaxation during the excited states in the fluorophores, the mechanisms involved are different. The former is related to charge transfer and less efficient recombination during the lifetime of the excited state, while the latter is directly related to rotational energy relaxation through the IC process, which transfers the energy from the siloles to the surroundings media. Actually, our designed experiments have successfully verify our hypothesis that AIE is attributed to restricted intramolecular low frequency rotations and excluded the likelihood of involvement of TICT and J-aggregate mechanism.
5 Applications
Light emitters are useful materials and have found a variety of technological applications. Great current interest is focused on the exploration of their utilities in the development of OLEDs and sensory systems. Many research groups have worked on the impediment of luminophore aggregation in an effort to fabricate efficient OLEDs, while other groups are developing luminescence turn-off sensors by utilizing the ACQ effect, in which the luminophores are incorporated into biological and chemical systems as labels via chemical attachment or probes via physical mixing. The labels/probes are initially dissolved and hence emissive (on) but become nonemissive (off) when they are induced to aggregate in the presence of analyte change.
The AIE-active siloles are highly emissive in the aggregate state. This unique characteristic differentiates them from the conventional fluorophores and makes them ideal candidates for high-tech applications in practically useful solid state. While many possibilities can be imagined for the AIE systems, we have mainly explored their applications as fluorescence sensors and in light-emitting devices.
5.1 Chemical Sensors
5.1.1 Vaporsensor
During our study on the AIE phenomenon, we have found that the emission of silole molecules on the TLC plate can be turned “off” and “on” by exposure to, and removal from, organic solvent vapors [34, 38]. Inspired by this fluorescence switch behavior, siloles are thus promising candidates to be used as organic vaporsensor. We investigated how the PL of MPPS would change upon solvent fumigation [38]. We dropped its solution onto TLC plates and put them in the petri dishes saturated with solvent vapor. As can be seen from Fig. 14, the emission from the MPPS spots is quenched when they are exposed to chloroform vapor. The solvent vapor may have condensed and hence formed thin liquid layers on the surfaces of the TLC plates, which has dissolved the absorbed MPPS molecules and thus turned “off” their light emission. When the solvent vapor is evaporated, the molecules aggregate and emit (“on”) again. This “off/on” process is completely reversible and can be repeated many times because of the nondestructive nature of the process. Such fluorescence switching behavior is also observed in other silole molecules, confirming that the vapochromism is a general phenomenon observable in the silole systems and suggesting that the silole molecules are promising candidates for fluorescence sensing of volatile organic compounds.

Photos of the spots of MPPS on the TLC plates in the petri dish sets (a) in the absence of and (b) after exposure to the vapor of chloroform solvent for about 1 min. Photo in (c) is taken in about 2 min after the solvent is evaporated. All the photographs are taken under UV illumination at room temperature
To have a spectroscopic picture, we followed the change in the PL of MPPS in the atmosphere saturated with organic vapors using a spectrofluorometer. A thin solid film of MPPS is coated on the inner wall of a quartz cell and several drops of acetone are added in a small container placed on the bottom of the cell. The emission of the silole film is then recorded at different time (Fig. 15a). Similar to the observation on the TLC plates, the PL of MPPS becomes weaker and is almost completely quenched after exposure to the vapor for ~2 min. However, the fumed film of HPS shows stronger and bluer emission than the untreated one (Fig. 15b), offering an opportunity to tune the vapochromism of siloles by changing their molecular structures [34].

Effect of acetone vapor on the PL spectra of films of siloles (a) MPPS and (b) HPS coated on quartz cells at different exposure times
5.1.2 Crystallization-enhanced Emission (CEE)
Why do siloles MPPS and HPS show different responses to the solvent vapor? It is known that solvent vapors can activate the dynamic recrystallization processes of many dyes. We thus investigated the morphologies of MPPS and HPS by SEM after solvent exposure. After solvent fuming, the MPPS film becomes smoother. Regularly shaped crystals are, however, formed in the fumed film of HPS. These results indicate that the organic vapor has dissolved the molecules in the MPPS film coated on the quartz cell and hence decreased their PL. On the contrary, the solvent vapor has helped the HPS molecules to crystallize in the supersaturated silole “solution” and thus shifted its PL spectrum to the bluer region with a higher intensity. The HPS molecules on the TLC plate are, however, well segregated by the fine silica gel particles, which make them difficult to crystallize. This accounts for the different behaviors of HPS on the TLC plate and the quartz cell. Silole 9 exhibits the same behavior as that of HPS. Since HPS and 9 possess a symmetric molecular structure, they may pack well during the fuming process. On the other hand, MPPS is asymmetric in shape, which may prevent it from packing into crystalline form under the solvent vapor atmosphere.
The above study shows that the silole crystals are stronger and bluer emitters than their amorphous counterparts. To confine whether this is the real case, we prepared MPPS crystals by recrystallization from an ethanol/THF solution and its amorphous powders by rapid quenching of its melt by liquid nitrogen. We investigated their structures by X-ray diffraction. As shown in Fig. 16, the crystals of MPPS give many sharp diffraction peaks at the low angles. Upon photoexcitation, the crystals emit a blue light at 460 nm. In contrast, the amorphous powders show broad halos of low intensities and are green emitters [38].

a Powder X-ray diffractograms and b photoluminescence spectra of crystalline and amorphous solids of MPPS
Crystallization usually red-shifts the emission spectra and decreases the emission intensity. Why is the PL enhanced and blue-shifted upon crystallization? As shown in Figs. 12 and 13, the silole molecules take propeller-shaped, instead of planar conformations due to the steric hindrance between the peripheral phenyl rings. The intramolecular distance is large, suggestive of the lack of strong molecular interactions that induces nonradiative recombination or red-shifts as observed in the “normal” crystals with strong π–π interactions. As the PL spectra of the amorphous aggregates are observed at longer wavelengths with lower intensities, this implies that the conformation in the amorphous state is not as twisted and rigid as that in the crystalline state.
5.1.3 Explosive Detection
Nitroaromatics such as 2,4-dinitrotoluene (DNT), 2,4,6-trinitrotoluene (TNT), and picric acid (PA) are warfare explosives and are important chemical species to detect in mine fields and munitions remediation sites. An example of PA detection using amine-containing silole 14 (A2HPS) is taken as follows [40]. The emission of the A2HPS nanoaggregates is weakened when PA is added into the suspension and is almost completely quenched in the presence of a large amount of PA (Fig. 17). The Stern–Volmer plot gives a quenching constant (K sv) of 1.67 × 105 M−1 with a R 2 value of 0.9930 in the [PA] range of 0–7.2 μM. Am- and HO-EPPSs also exhibit a very high sensitivity to the PA quenching [42], with respective Stern–Volmer constants as large as 1.1 × 105 and 8.0 × 104 M−1. The high sensitivity of these siloles to PA detection makes them promising candidates as sensors for future real-world applications.

a PL spectra of A2HPS (14) in THF/H2O mixtures (100:1 by volume) with different concentrations of picric acid. b Plot of PL intensity versus PA concentration. c Linear region of the (I 0/I − 1)–[PA] plot in panel b
5.1.4 pH Sensing
A2HPS is insoluble in water. However, after adding a small amount of sulfuric acid, A2HPS dissolves readily because its tertiary amino groups are transformed into ammonium salts to give H2A2HPS2+ [40]. The aqueous solution is nonluminescent because the salt molecules are genuinely dissolved in the acidic medium. What will happen if the acid is neutralized by an aqueous potassium hydroxide solution? The emission intensity remains unchanged when the pH value is increased from 2 to 5.4 but starts to swiftly increase afterward (Fig. 18) [40]. At a pH value of 6.35, the emission is >150-fold stronger than that at pH = 2. This is easy to understand. At low pH values, the dye molecules exist in their ammonium salt form and are thus soluble in water. When the pH value exceeds 5.4, the dye molecules are converted to their amine forms. The decrease in the hydrophilicity induces the molecules to aggregate in the aqueous medium, and thus turns on their emissions. This reversible switch “off” and “on” at low and high pH values due to the ionization and deionization under acidic and basic conditions, respectively, makes A2HPS an excellent pH sensor. HO-EPPS can also work as a pH sensor in an exact opposite direction because the ionization of its hydroxy substituent may enable it to dissolve in an aqueous medium at high pH values or under basic conditions [42].

a A2HPS and its protonated form H2A2HPS2+. b Emission spectra of A2HPS in aqueous solutions with different pH values and c effect of pH on the emission of A2HPS. Inset: Photographs of A2HPS taken under illumination of a UV lamp at pH values of 2 and 12
5.1.5 Monitoring Layer-by-Layer (LbL) Self-assembling
LbL deposition of polyelectrolytes has been widely used to construct functional thin solid films. UV–vis absorption has been used to monitor the assembling process. The method, however, requires that the polyelectrolytes carry chromophores (e.g., polystyrene) and that the substrates are transparent (e.g., quartz). The LbL deposition is basically a polymer aggregation process, which thus offers a stage for an AIE-active siloles to perform as an in situ and real-time monitor.
When a mixture of poly(diallyldimethylammonium chloride) (PDDAC) and H2A2HPS2+ is used as a co-cation to assemble with poly(styrene-sulfonate) (PSS) anion by the LbL deposition process on a quartz plate, the emission intensity of H2A2HPS2+ is linearly increased with increasing number of the bilayers (Fig. 19) [63]. The same linear calibration curve is obtained when inexpensive glass plate is used as substrate. The AIE luminogen is not limited to the examples given in Fig. 19 but can in principle be used as a probe for monitoring assemblies of polyelectrolytes free of chromophores on any substrates, including the technologically useful silicon wafer and mica.

a PL spectra of [(H2A2HPS2+ + PDDAC)/PSS] n films with different number of bilayers (n) deposited on a quartz plate. Abbreviation: PDDAC = poly(diallyldimethylammonium chloride), PSS = poly(styrenesulfonate). b Plots of PL intensities versus numbers of bilayers deposited on quartz and glass plates
5.1.6 Detection of Critical Micelle Concentration (CMC)
Micelles are ubiquitous in nature. They have attracted considerable research attentions due to their multiple functions as entities of emulsion polymerization [64–66], templates for synthesizing nanomaterials [67–70], and controllable drug delivery [71–76]. Varieties of natural and synthetic amphiphilic molecules and macromolecules have been derived to build micelles for specific applications. The determination of the CMC is, however, a fundamental task. A few methods, e.g., osmotic pressure, equivalent conductivity, and surface and interfacial tension, have been established to measure the CMC values. These methods depend on professional instruments and thereby are time-consuming and expensive. Recently, we have developed a facile, efficient, and unprecedented method for CMC measurement using AIE-active siloles. The hexaphenylsiloyl ring in H2A2HPS2+ is hydrophobic, while its ionized ammonium groups are hydrophilic; in other words, the molecule is amphiphilic. When it dissolves in aqueous surfactant solutions with different concentrations at low pH values, the following phenomenon is observed (Scheme 11). Faint PL will be recorded when the concentration of the surfactant is lower than the CMC because both H2A2HPS2+ and surfactant molecules are dissolved in water. Once the concentration comes to or becomes higher than the CMC, the H2A2HPS2+ molecules will enter and aggregate in the hydrophobic cores of the micelles. Thus, the sudden increase in PL signal may indicate the formation of micelles.

Illustration of CMC detection based on the AIE mechanism
Figure 20 shows the PL spectra of H2A2HPS2+ in buffer solutions (pH = 4) containing different concentrations of cationic surfactant cetyltrimethylammonium bromide (CTAB) [77]. When the concentration of CTAB is lower than 0.1 mg/mL, no significant change in the PL intensity is observed. At a concentration of 0.3 mg/mL, the PL intensity rises abruptly. Further increasing the surfactant concentration to 5 mg/mL, the PL intensity grows much higher. Plotting the PL intensity against the CTAB concentration gives two lines that intersect at a concentration of ~0.23 mg/mL, which is the CMC value for the micellar system. Before the inflexion, the PL intensity suffers little change with the CTAB concentration. Afterwards, the intensity elevates monotonously. Form the photographs shown in Fig. 20c, we can clearly see the critical PL intensity change at ~0.3 mg/mL. The literature CMC value for CTAB is 0.3 mg/mL. Thus, the experiment result reported here is well consistent with the accredited data [78]. This method has been successfully applied to CMC detection of cationic and anionic surfactants, amphiphilic biomolecules, and block copolymers. Herein, we choose biomolecule lecithin as an example [79]. When the concentration of lecithin is low, it is molecularly dissolved. The molecules of H2A2HPS2+ are also genuinely dissolved in the acidic medium and thus the solution is nonemissive. When large amounts of lecithin are present in the solution of H2A2HPS2+, the solution becomes emissive. It is because at CMC, the lecithin molecules start to form micellar nanoaggregates. When the H2A2HPS2+ molecules aggregate in the micelles, they start to emit, as indicated from the photographs in Fig. 21. The AIE silole thus functions as a handy nanoaggregate beacon for CMC evaluation.

a PL spectra of buffer solution (pH = 4) of H2A2HPS2+ with different concentration of CTAB. b Plot of emission peak intensity versus [CTAB]. c Photographs of H2A2HPS2+ in aqueous solutions with different [CTAB] were taken under illumination of a handheld UV lamp

Plot of PL intensity of H2A2HPS2+ versus concentration of lecithin in an acidic aqueous medium (pH = 3). Photographs of the solutions were taken under illumination of a handheld UV lamp
5.2 Biological Probes
5.2.1 Detection of Biomacromolecules
We have succeeded in utilizing H2A2HPS2+ as a bioprobe for detecting biomacromolecules (Fig. 22) [40]. The buffer solution (pH = 2) of H2A2HPS2+ emits only a faint light. Upon addition of bovine serum albumin (BSA), the dye solution becomes emissive. The PL intensity rises with an increase in BSA concentration and a 52-fold emission enhancement is achieved at a concentration of 500 μg/mL. Thus, H2A2HPS2+ is an excellent “light-up” biosensor for the protein detection.

a PL spectra of H2A2HPS2+ in buffer solutions (pH = 2) containing different amounts of BSA and b plot of PL intensity versus BSA concentration. Inset: photographs of H2A2HPS2+ in buffer solutions without and with 500 μg/mL of BSA taken under illumination of a handheld UV lamp. c PL spectra of H2A2HPS2+ in buffer solutions (pH = 2) containing different amounts of hs DNA and d plot of PL intensity versus hs DNA concentration. Inset: photographs of H2A2HPS2+ in buffer solutions without and with 200 μg/mL of hs DNA taken under illumination of a handheld UV lamp
The effect of DNA is even more pronounced. The emission of H2A2HPS2+ is turned on when herring sperm (hs) DNA is added to its buffer solution. The intensity is monotonically increased with an increase in the DNA concentration. At the same concentrations, the (I/I 0 − 1) values are higher than those induced by BSA, indicating that H2A2HPS2+ is a more sensitive probe for DNA detection. The linear range of I/I 0 − 1 versus [hs DNA] plot is as wide as 0–100 μg/mL, with a correlation coefficient as high as 0.997.
Similarly, HO-EPPS can be used as a probe for RNAs by virtue of its AIE nature (Fig. 23) [42]. Progressive addition of RNA R6625 into alkalified HO-EPPS in the methanol/water mixtures leads to monotonical increase in the PL intensity. Interestingly, the emission enhancement in the case of RNA R6625 is bigger than that of RNA R3629.

a PL spectra of HO-EPPS (24) in the methanol/water mixtures (4:6 v/v) alkalified by KOH in the presence of varying amounts of RNA R6625; The spectrum of a RNA R6625 solution is shown in dashed line for comparison. b Plots of emission intensities of HO-EPPS at 503 nm versus concentrations of different RNAs
In the acidic buffer solutions containing the biomacromolecules, the cationic amphiphilic dye molecules may bind to the negatively charged DNA and RNA or enter into the hydrophobic pockets in the native folding structure of the protein via electrostatic attraction and hydrophobic effect. When they are docked on the surfaces of the biomacromolecules or located in the cavities of their folding structures, their intramolecular rotations of the dye molecules are suppressed, which impedes the nonradiative decays and populates the radiative transitions.
5.2.2 Visualizing Agent for Gel Electrophoresis
Gel electrophoresis is a technique used for the separation of DNA, RNA, or protein molecules using an electric field applied to a gel matrix. After the electrophoresis is completed, the separated analytes in the gel can be stained by fluorescent dyes to make them visible under UV light. Ethidium bromide (EB) is commonly used as a nucleic acid stain for agarose gel electrophoresis in molecular biology laboratories. When exposed to UV light, it emits a red-orange color, which intensifies almost 20-fold after binding to DNA. However, EB can be intercalated into DNA. Thus, it may be a very strong mutagen, and may possibly be a carcinogen or teratogen. If we can find another dye with low toxicity and cost to replace EB, it will be a great achievement in the development of biological techniques.
As mentioned above, siloles will emit more strongly when bound to biomacromolecules. Such behavior stimulates us to use siloles as staining agents for biomacromolecules. Normally, EB is preloaded in the agarose gel before electrophoresis, while we used buffer solution of silole 15 to stain agarose gel after electrophoresis. Gel electrophoresis of two DNA ladder markers M1 and M2 is carried out using EB and 15 as nucleic acid stains for comparison. Although the results shown in Fig. 24 are preliminary, it can be seen that 15 is promising stain for visualization of DNA with different sizes. We are now designing and testing different siloles to optimize the conditions.

Gel electrophoresis of DNA ladder markers M1 and M2 using (a) ethidium bromide (EB) and (b) silole 15 as nucleic acid stains
5.2.3 Biolable for Immunoassay
In the traditional fluorescence immunoassay (FIA) systems, the fluorophore to protein (F/P) ratios are often low because of the problems caused by the notorious self-quenching effect. When several fluorophoric labels are attached to one antibody, the fluorophores are located in close vicinity, which activates energy transfer and decreases the PL intensity and efficiency. The AIE effect permits the use of high F/P ratios and the AIE fluorophores can thus serve as powerful immunosensors. Bio-functionalized HPS nanocrystals are used in our FIA study, which are prepared by the procedures shown in Fig. 25a. The HPS crystals are ball-milled in a mixture of hydroxypropyl cellulose and SDS in water (step 1). After encapsulation with polyelectrolyte multilayers (step 2), specific immunoreagents such as antibodies are attached to the nanocrystals (step 3). The nanocrystal core is comprised of a huge number of HPS molecules, while the encapsulated crystal surface is decorated with biomolecules. This configuration confers an extremely high F/P ratio on the AIE immunosensor.

a Schematic illustration of a sandwich-type immunoassay process using polyelectrolyte-encapsulated, antibody-functionalized HPS nanocrystal as fluorescent bioprobe. b Plot of PL intensity of the HPS nanocrystal functionalized by goat anti-mouse immunoglobulin G versus the concentration of mouse immunoglobulin G (M IgG). Data for the system using fluorescein isothiocyanate (FITC) as bioprobe is shown for comparison
The nanocrystalline HPS biomarkers are used for sandwich-type immunoassays [29]. The analyte is first immobilized by the capture antibody pre-adsorbed on a solid substrate and then exposed to the antibody-labeled bioprobe (step 4). The PL intensity is increased with the analyte concentration (Fig. 25b). The sensitivity of the HPS bioprobes is 140-fold higher than that of fluorescein isothiocyanate (FITC)–antibody conjugate. The signals of the nanocrystalline bioprobes are dramatically amplified, thanks to the extremely high F/P ratios in the FIA system. This manifests the great value of the AIE effect to the development of ultrasensitive FIA systems.
5.2.4 Cell Imaging
Biocompatibility is the key factor that determines whether a biosensor can be used for in vivo applications. To be a useful cell tracer, a fluorogen should neither inhibit nor promote the growth to living cells. Cytotoxicity of 63 is tested in dimethyl sulphoxide (DMSO) solutions with a DMSO content of <0.1%, as DMSO exerts no biological effect on the cell proliferation when its amount in the medium is <0.2% [80]. In the presence of 63, the HeLa cells grow as normal as they do in the control experiment (Fig. 26). Evidently, 63 is nontoxic to the living cells. In other words, it is cytocompatible without interfering with the metabolisms of the living cells.

Effect of silole derivative 63 on viability of living HeLa cells evaluated by MTT assay. MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide
The excellent biocompatibility of 63 prompted us to utilize it for intracellular imaging [80]. When the HeLa cells are stained by the nanoaggregates of 63, the cells are clearly imaged in an exposure time as short as 1 s (Fig. 27a). Under the identical imaging conditions, commercial fluorescent dye CellTracker™ Green CMFDA shows an inferior performance: the images of the cells can hardly been seen when the stained HeLa cells are exposed for 1 s (Fig. 27b). Clear images can finally be taken when the exposure time is prolonged to 5 s.

Fluorescence images of living HeLa cells stained by (a) 63 and (b, c) CellTracker™ Green CMFDA. Exposure time: (a, b) 1 s, (c) 5 s
Closer inspection of the fluorescence images reveals that the nanoaggregates of 63 stain the cytoplasmic regions of the cells but the CMFDA molecules stain the entire cells. This is probably because the former is hydrophobic but the latter is hydrophilic. Water-soluble fluorophores such as CMFDA can pass through membranes to enter the cells. The small CMFDA molecules can also enter the nucleus through nuclear pores. The fluorophore therefore can label both the cytoplasmic and nuclear compartments of the living cells (Fig. 27c).
The major route for the nanoaggregates of 63 to enter a cell is through endocytosis. During this process, the aggregates are enclosed by the cell membrane to form small vesicles that can be internalized by the cell. Inside the cell, the nanoaggregates can be further processed in endosomes and lysosomes and are eventually released from the cellular organelles. However, the hydrophobic nature of the nanoaggregates prevents them from entering the nucleus of the cell. When the nanoaggregates are bound to the biopolymers in the cytoplasm, they become very luminescent, due to the additional physical restriction to their intramolecular rotation.
The fact that the nanoaggregates of 63 selectively stain the cytoplasmic regions of the cells gives this AIE fluorogen a unique advantage over the CellTracker that stains the whole cells. In most cases of cell imaging, it is necessary to use two different fluorogens to stain a cell: one to stain DNA in the nucleus, while another to stain the cytoplasm. Our AIE dye is thus a better choice when used in combination with a DNA-staining fluorogen.
5.2.5 Fluorescent and Magnetic Nanoparticles
Fluorescent silica nanoparticles (FSNPs) can provide high fluorescent intensity compared to the common organic fluorescent dyes, which may find high-tech applications in ultrasensitive assay, biological imaging, etc. Normally, FSNPs contain conventional dyes, e.g., FITC and RITC, inside of the silica matrix. The incorporated dyes gain additional photochemical stability even after multiple exposures to light, thus enabling long-term cell imaging studies. However, their fluorescence is often weak [17, 81–85]. This is due to the emission quenching caused by the aggregation of the dye molecules in the solid state. A low dye loading in the particle may be free of aggregation but can only offer weak emission. The light emission will be weakened by loading more dye molecules into a particle because of the notorious ACQ effect. Much effort has been devoted to the mitigation of aggregate formation of dye molecules in the solid state, which has, however, met with only limited success [86–88]. Also, a magnetic material, e.g., Fe3O4, can be inserted into the core of nanoparticles for offering magnetic properties. The silica shell surrounds the magnetic Fe3O4 core and contains organic dyes inside, so that it can exhibit both magnetic and optical properties. Moreover, the outer surface of the silica shell is easily modified with functional ligands for specific biological applications.
Instead of quenching, aggregation has enhanced the light emission of siloles. Thus, they are ideal dyes for the fabrication of fluorescent and magnetic nanoparticles. We prepared silole-doped fluorescent SiO2 and magnetic fluorescent Fe3O4@SiO2 core-shell nanoparticles according to the procedures shown in Scheme 12. The fluorescent SiO2 nanoparticles are uniform in size (panels a and b of Fig. 28). While the solution of 66 and the pure silica nanoparticles are nonemissive, the 66-doped nanoparticles are highly fluorescent under UV illumination (Fig. 28b, inset). We have applied these nanoparticles for cell imaging as shown in panels c and d of Fig. 28, which selectively stain the cytoplasmic regions of the cells. Importantly, we have successfully prepared 66-doped Fe3O4@SiO2 core-shell nanoparticles with a uniform size of ~50 nm, which enjoy both fluorescent and magnetic [saturation magnetization (M s) ~6 emu/g] properties (panels a and b of Fig. 29). They can be reversibly collected and dispersed in solutions by switching on and off the magnetic field, which may be potentially used for simultaneous bio-imaging and -separations. These nanoparticles can be internalized into the live cells by endocytosis and provides a long-term photostability for fluorescent imaging and dynamic studies. It is also available to get an in vivo imaging by magnetic resonance imaging (MRI) technique without giving damage to the animal. As the preliminary results shown in panels c and d of Fig. 29, the magnetic fluorescent nanoparticles can enter the cells and only lighten the cytoplasmic regions of the HeLa cells. Our research group is actively working on modifying outer surfaces of the nanoparticles with functional ligands for specific biological applications by using fluorescence and MRI techniques.

Syntheis of silole-loaded fluorescent silica nanoparticles and magnetic fluorescent Fe3O4@SiO2 core-shell nanoparticles

a SEM and b TEM images of fluorescent silica nanoparticles loaded with silole 66; inset of panel b: nanoparticles homogeneously dispersed in ethanol under UV illumination. Fluorescent microscopy images of living HeLa cells stained by fluorescent silica nanoparticles: c phase contrast images and d fluorescence images

a Plot of magnetization (M) versus applied magnetic field (H) at 300 K for the 66-doped magnetic fluorescent core-shell Fe3O4@SiO2 nanoparticles. Inset: TEM images of the nanoparticles. b PL spectra of 66-doped Fe3O4@SiO2 and pure Fe3O4 nanoparticles. Inset: photographs showing reversible dispersion and collection of nanoparticles in solution by switching the magnetic field under UV illumination. c and d Phase contrast images and fluorescent microscopy images of living HeLa cells stained by the 66-doped magnetic fluorescent core-shell Fe3O4@SiO2 nanoparticles
5.3 Stimuli-responsive Organic Nanomaterials
As described above, the nanoaggregates of siloles suspended in the aqueous mixtures can be used as fluorescent chemosensors. We proceeded further to develop solid-state detection strips for chemical species such as metallic ions in water, which may find applications in water quality control, pollution tracing, and environmental protection [89]. Employing an electrospinning technique, composite films are prepared from the blends of HPS and PMMA. Macroscopically, the thin films look uniform but microscopically they are found to contain many microspheres and nanofibers (Fig. 30a). There exist many nanosized pores and protuberances in the microspheres. This distinct structure resembles that of a lotus leaf surface. The contact angle (CA) of a water droplet on the HPS/PMMA micro/nanocomposite film is 115 ± 2.8o, indicating that the film surface is very hydrophobic (Fig. 30a, inset).

a SEM image of electrospun HPS/PMMA composite film. Inset: photograph of the shape of a water droplet on the film with a contact angle of 115 ± 2.6o. b PL spectra of the electrospun film in pure water (HPS/PMMA) and in an aqueous Fe3+ solution (HPS/PMMA + Fe3+). Inset: reversible switching between emissive and nonemissive states by immersing the HPS/PMMA film into pure water and Fe3+ solution, respectively
In pure water, the film emits a strong blue green light at 481 nm (Fig. 30b). This emission is quenched when the film is immersed in an aqueous ferric solution, giving a K sv value of 6.21 × 103 M−1. The Fe3+ ions can be washed away by placing the film in pure water, thanks to its lotus-like structure and high hydrophobicity. By alternatively putting the film in pure water and ferric solution, the emission can be repeatedly and reversibly switched between bright and dark states with excellent reproducibility for many cycles (Fig. 30b, inset).
Fabrication of nanowires has attracted much interest owing to their unique materials properties and wide practical applications. Nanowires of HPS can be facilely prepared by using anodic aluminum oxide (AAO) membrane as template [39, 90]. Examples of HPS nanowires with average diameters of 70 nm and 150 nm are shown in panels a and b of Fig. 31, respectively. The nanowires are aligned in a continuous, parallel manner with an average length of ca. 60 μm. The nanowires are highly luminescent due to the AIE property of the silole aggregates. Intriguingly, their emission peaks bathochromically shift with an increase in their diameters (Fig. 31c) [39].

SEM images of HPS nanowires with average diameters (d) of a 70 and b 150 nm. c PL spectra of HPS nanowires with average diameters of 35 and 250 nm. Inset: plot of emission maximum (λ max) versus nanowire diameter (d)
In our early studies, we have observed that the emissions of AIE aggregates bathochromically shift when they undergo a morphology change from crystalline to amorphous state (vide supra). Crystals of HPS, for example, emit a blue light of ~465 nm, while its amorphous powders emit a green light of ~490 nm. The emission peak of the “thin” HPS nanowires (d = 35 nm) is close to that of the HPS crystals, suggesting that these nanowires are crystalline in morphology. On the other hand, the “thick” nanowires (d = 250 nm) emit at 495 nm, close to that of the HPS powders (Fig. 31c). This indicates that the red shift in the emission peak with increasing nanowire size stems from the crystalline to amorphous phase transition.
In the nanowire fabrication process, the nanopores in the AAO template are filled with saturated HPS solution due to the capillary action. The saturated vapor tension in the small channels (e.g., 35 nm) is high, which slows down the solvent evaporation process. This gives the HPS molecules enough time to reorient and crystallize. As the diameter of the AAO channel is increased, the saturated vapor tension is decreased. This accelerates the solvent evaporation, giving no time for the HPS molecules to pack regularly but agglomerate abruptly. Amorphous nanowires are thus formed in the AAO channels with large diameters. Evidently, the PL of the nanowires can be tuned by changing their morphologies, which are realized by simply varying the channel sizes of the AAO template.
“Smart” or “intelligent” materials change their properties in response to external stimuli or perturbations. Most of the existing stimuli-responsive smart materials change a single property in a “one-way” fashion. The research of “two-way” intelligent materials, i.e., those capable of reversibly switching between two properties, is still in its infancy. We have developed a dual-responsive system based on HPS [41]. The film prepared by spin-coating is smooth and featureless (Fig. 32A), suggesting that the film is amorphous. Fuming the film by ethanol vapour induces the HPS molecules to reorient, giving a crystalline film full of fine structures of nanosheets and nanorods (Fig. 32B). This morphological change is reversible: the crystalline film can be transformed to the amorphous one by exposing it to toluene vapour (Fig. 32C).

A Amorphous and B crystalline films of HPS. C Shapes of water droplets on, and colors of light emissions from, the amorphous and crystalline films of HPS. D Reversible switching of contact angle and emission color of HPS by repeated exposure of its amorphous and crystalline films to ethanol and toluene vapors, respectively
The surface wettability of the thin films is evaluated by CA measurement. The CA of the amorphous film is 97.0 ± 1.5o, indicating that its surface is hydrophobic. After controlled fumigation by ethanol vapour, the water droplet shrinks to a CA value of 136.3 ± 1.6o, meaning that the film becomes more hydrophobic (Fig. 32C). The crystalline film is changed to the amorphous one when fumed by toluene vapour, accompanying with a drop in the CA value (from 136o to 97o) and a shift in the PL color (from blue to green). The changes in the surface wettability and solid-state emission can be repeated many times without fatigue, indicative of an excellent reversibility of the two-way switching processes (Fig. 32D).
5.4 Photoelectronic Devices
5.4.1 OLED
OLEDs have attracted a great deal of attentions because of their potential application as new display devices [91–95]. To achieve high EL efficiency in OLEDs requires balanced injection and transportation of holes and electrons, and chromophores with efficient PL in the solid state. Charge injection can be equalized either by using metals with low work functions as cathodes or by adjusting the HOMO/LUMO levels of the organic layer. However, it is difficult to balance the charge transportation because of the inherent hole-transporting properties of organic semiconducting materials. Electron transporting (ET) organic materials with high mobility are rare and in great demand. Conventional organic semiconductors suffer from ACQ effect originating from the formation of excimer or exciplex in the aggregation state, which decreases their PL efficiency. The ACQ effect has been a thorny problem in the development of efficient OLED because chromophoric molecules are normally used as thin solid films in the EL devices. Although both hole and electron mobilities are drastically increased by changing the film morphology from amorphous to crystal state, the ACQ effect will be intensified. Such self-contradiction is difficult to handle.
The novel AIE and CEE features of siloles really make them promising candidates as EL materials. Aggregation including crystallization can be a positive factor to improve the performances of EL devices. Moreover, siloles are excellent electron-transporting materials [36]. An electron mobility of 2.1 × 10−6 cm2/V s in the MPPS film at an electric field of 1.2 × 106 V/cm is obtained. This mobility is comparable to that of AlQ3 (2.3 × 10−6 cm2/V s) [96], which is one of the most extensively used electron transport materials in OLEDs, at the same electric field. The electron mobility of the HPS film is about ~1.5 times higher than that of the MPPS film. Recently, we have made siloles with higher electron mobilities (cf., Fig. 33). By using the transient EL method, the electron mobilities of 70b and 70a were found to lie in the ranges of 5.0 × 10−6–1.2 × 10−5 and 3.1–6.3 × 10−6 cm2/V s, respectively, which are higher than that of AlQ3 (2.3–4.3 × 10−6 cm2/V s at electric fields ranging from 1.2 to 4.0 × 106 V/cm). It is known that AlQ3 exhibits a very low ΦF in the solid state, while siloles possess much higher ΦF’s. We fabricated silole-based LEDs and obtained outstanding performances. The best efficient devices are constructed with MPPS and HPS, which exhibit high current and power efficiencies (Fig. 34). The external quantum efficiencies (ηEL) are 8% and 7%, for MPPS and HPS, respectively, approaching the limit of the possible in organic singlet emitters [25, 26]. Therefore, siloles are a group of excellent emissive and ET materials for OLED application. These outstanding properties are originated from their intrinsic propeller-shaped structures and high electron affinity arising from σ*–π* conjugation [7, 36], which decrease the probability of the excimer or exciplex formation and enhance the electron mobility. To the best of our knowledge, silole is one of the most emissive materials that possess both high charge carrier mobility and PL efficiency in the solid states.

Electron mobility of MPPS, HPS, 70a, and 70b plotted as a function of the square root of a bias field

Configuration of silole-based LEDs and EL performances of MPPS and HPS. Abbreviations: V on = turn-on voltage; CE = current efficiency; PE = power efficiency; L = luminance; ηEL = external quantum yield
5.4.2 Photovoltaic Cell
The high electron affinity of siloles has spurred our interest to utilize them as active materials to construct PV cells. It has been well recognized that excitons dissociate efficiently at a donor (D)–acceptor (A) heterojunction interface [97–107]. Carbazole (Cz) is a well-known electron donor and widely used hole transport material. We envisioned that introduction of Cz donor groups into the silole acceptor system may create photo-responsive D–A adducts. Under the influence of an applied bias, the dissociated holes and electrons in the respective D and A domains may steadily migrate along the interfaces to corresponding electrodes to finish the PV process of converting light to electricity. As can be seen from Fig. 35, all of the PV cells of Cz2HPS (17) (devices I–VI) show good PV efficiencies [35]. The best results are obtained with device III, whose short-circuit current density, open-circuit voltage, and fill factor are 96.5 μA/cm2, 1.7 V, and 0.21, respectively. Although the structure of the cell is far from optimized, it already shows an ηPV as high as 2.19%. Optimization of the device structure may further boost the ηPV of the Cz2HPS-based PV cell.

I–V characteristics of PV cells of Cz2HPS (17)
5.4.3 Polarized Emission
Recently, considerable synthetic and processing efforts have been devoted to developing highly polarized fluorescence materials for photoelectronic display devices. However, most of them show weak fluorescence in the solid state. If a material can possess a high fluorescence emission efficiency and emission anisotropy, it will be a promising candidate for advanced photoelectronic devices. During our study, we found that 70c hardly crystallized but exhibited an excellent one-dimensional self-assembling ability. By adding CH3OH, a relatively poor solvent of 70c, to its CH2Cl2 solution (1 mg/mL) in a glass tube, fabirls with nanometer sizes are readily formed. With slow pervasion of CH3OH and solvent evaporation, the solution is concentrated and more nanofibrils are formed and self-assemble into microfibers. Figure 36 shows the SEM, TEM, and fluorescence microscopy images of the microfibers. It can be seen that the microfibers comprise nanofibrils with an average diameter of about 50 nm and a length of several tens of micrometers. It is envisioned that these highly fluorescent microfibers may have polarized emission. As expected, emission anisotropy was observed in microfibers of 70c. The emission intensity of the fibrous aggregates is stronger in the vertical direction (I v) than in the horizontal direction (I h) (Fig. 37a), probably due to its rigid molecular conformation and better alignment in the microfibers. The conformational stiffening and fluorogenic orientation should be further enhanced in the crystalline state as the molecules are confined in the crystalline lattice. The polarization (P) and emission anisotropy (r) thereby should be larger in the crystals than those in the microfibers. Indeed, I v is much higher than I h in the crystals of 70a (I v/I h ~ 4.5) (Fig. 37b). Inspired by these excellent attributes, viz., polarized fluorescence emission and high emission efficiency in solid state, we are now actively exploring their applications for advanced light-emitting devices.

(a and b) SEM, (c and d) TEM, and (e and f) fluorescence microscopy images of microfibers of 70c consisting of nanofibrils. Insets: enlarged images [(b) and (f)] and electron diffraction pattern (d)

Polarization emissions in the (a) fibrous aggregates of 70c and (b) crystals of 70a. Degree of polarization (P) and emission anisotropy (r) are determined by P = (I v − I h)/(I v + I h) and r = (I v − I h)/(I v + 2I h), where I v and I h are the PL intensities at vertical and horizontal directions, respectively
6 Conclusions
A novel class of silole molecules and their polymers with AIE property is developed. This “abnormal” but excellent AIE property is attributed to restricted intramolecular rotations of phenyl rings against the central silole core. This hypothesis is verified by our designed experiments, which help exclude the likelihood of involvement of TICT or J-aggregate mechanism. The silole solutions are weakly luminescent or nonluminescent because the free rotations of phenyl rings around the central silole core relax the energy of the excited states and transfer it to the surrounding solvent molecules. External perturbation on the silole solutions by adding poor solvent, increasing solvent viscosity, or decreasing solution temperature has boosted their ΦF’s by up to two orders of magnitude. Pressurization increases the PL intensity of siloles in the solid state. Internal structural perturbation by molecular engineering endeavor can also enhance their emission efficiency. All the imposed stimuli restrict the intramolecular rotations of peripheral aromatic rings, which block the nonradiative channel and effectively populate the radiative decay of the excitons.
We utilized this valuable AIE property to explore the applications as: sensors for chemicals, explosives, pH, and biomacromolecules (proteins, DNAs and RNAs), indicators for determining CMC and monitoring layer-by-layer self-assembling, biocompatible fluorogens for cell imaging, visualizing agent for DNA gel electrophoresis, biolabels for immunoassay, stimuli-responsive organic nanomaterials, magnetic fluorescent nanoparticles for potential bio-imaging and separation, and excellent materials for efficient OLEDs and PV cells. Especially, external quantum yields of 8% and 7% are achieved in MPPS- and HPS-based OLEDs, respectively, which show the apparent violation of the upper theoretical limit of 5.5% for a singlet emitter.
The discovery of AIE property really refreshes our knowledge about aggregation effect on the photophysical process. Aggregation is not always a negative factor for the light emission of luminophores. In siloles, aggregation including crystallization has played a positive role to enhance their emissions, which is particularly helpful for the development of efficient EL devices, as demonstrated by the outstanding performances of silole-based OLEDs. We believe that the AIE phenomenon occurs in many other molecular systems and this exciting but insufficiently exploited field is waiting for scientists to carry forward and develop new photophysical theories to direct technological innovations in the near future.
References
E.H. Braye, W. Hübel, Chem. Ind. (London) 1250 (1959)
E.H. Braye, W. Hübel, I. Caplier, J. Am. Chem. Soc. 83, 4406 (1961)
J. Dubac, A. Laporterie, G. Manuel, Chem. Rev. 90, 215 (1990)
E. Colomer, R.J.P. Corriu, M. Lheureux, Chem. Rev. 90, 265 (1990)
A.P. Sadimenko, Adv. Heterocycl. Chem. 79, 115 (2001)
V.Y. Lee, A. Sekiguchi, M. Ichinohe, N. Fukaya, J. Organomet. Chem. 611, 228 (2000)
K. Tamao, M. Uchida, T. Izumizawa, K. Furukawa, S. Yamaguchi, J. Am. Chem. Soc. 118, 11974 (1996)
S. Yamaguchi, K. Tamao, J. Chem. Soc., Dalton Trans. 3693 (1998)
B. Wrackmeyer, Coord. Chem. Rev. 145, 125 (1995)
C. Chuit, R.J.P. Corriu, C. Reye, J.C. Young, Chem. Rev. 93, 1371 (1993)
H. Murata, G.G. Malliaras, M. Uchida, Y. Shen, Z.H. Kafafi, Chem. Phys. Lett. 339, 161 (2001)
H. Murata, Z.H. Kafafi, M. Uchida, Appl. Phys. Lett. 80, 189 (2002)
J. Ohshita, H. Kai, A. Takata, T. Iida, A. Kunai, N. Ohta, K. Komaguchi, M. Shiotani, A. Adachi, K. Sakamaki, K. Okita, Organometallics 20, 4800 (2001)
C. Hay, M. Hissler, C. Fischmeister, J. Rault-Berthelot, L. Toupet, L. Nyulaszi, R. Reau, Chem. Eur. J. 7, 4222 (2001)
S. Yamaguchi, T. Endo, M. Uchida, T. Izumizawa, K. Furukawa, K. Tamao, Chem. Eur. J. 6, 1683 (2000)
J.B. Birks, Photophysics of Aromatic Molecules (Wiley, New York, 1970)
B.K. An, S.K. Kwon, S.D. Jung, S.Y. Park, J. Am. Chem. Soc. 124, 14410 (2002)
C.J. Bhongale, C.S. Hsu, Angew. Chem. Int. Ed. 45, 1404 (2006)
M.R. Han, M. Hara, New J. Chem. 30, 223 (2006)
C. Yuan, X. Tao, Y. Ren, Y. Li, J. Yang, W. Yu, L. Wang, M. Jiang, J. Phys. Chem. C 111, 12811 (2007)
H. Wang, Z. Xie, Y. Ma, J. Shen, Sci. China Ser. B 50, 433 (2007)
J. Chen, Y. Cao, Macromol. Rapid Commun. 28, 1714 (2007)
Y.T. Lee, C.L. Chiang, C.T. Chen, Chem. Commun. 217 (2008)
J. Luo, Z. Xie, J.W.Y. Lam, L. Cheng, H. Chen, C. Qiu, H.S. Kwok, X. Zhan, Y. Liu, D. Zhu, B.Z. Tang, Chem. Commun. 1740 (2001)
H. Chen, W.Y. Lam, J. Luo, Y. Ho, B.Z. Tang, D. Zhu, M. Wong, H.S. Kwok, Appl. Phys. Lett. 81, 574 (2002)
J. Chen, C.C.W. Law, J.W.Y. Lam, Y. Dong, S.M.F. Lo, I.D. Williams, D. Zhu, B.Z. Tang, Chem. Mater. 15, 1535 (2003)
J. Chen, Z. Xie, J.W.Y. Lam, C.C.W. Law, B.Z. Tang, Macromolecules 36, 1108 (2003)
J. Chen, H. Peng, C.C.W. Law, Y. Dong, J.W.Y. Lam, I.D. Williams, B.Z. Tang, Macromolecules 36, 4319 (2003)
C.P.Y. Chan, M. Haeussler, B.Z. Tang, Y. Dong, K.K. Sin, W.C. Mak, D. Trau, M. Seydack, R. Renneberg, J. Immuno. Methods 295, 111 (2004)
Y. Ren, J.W.Y. Lam, Y.Q. Dong, B.Z. Tang, K.S. Wong, J. Phys. Chem. B 109, 1135 (2005)
Y. Ren, Y. Dong, J.W.Y. Lam, B.Z. Tang, K.S. Wong, Chem. Phys. Lett. 402, 468 (2005)
Z. Li, Y.Q. Dong, B. Xiu, Y. Tang, M. Haeussler, H. Tong, Y.P. Dong, J.W.Y. Lam, Y. Ren, H.H.Y. Sung, K.S. Wong, P. Gao, I.D. Williams, H.S. Kwok, B.Z. Tang, J. Phys. Chem. B 109, 10061 (2005)
J. Chen, B. Xu, K. Yang, Y. Cao, H.H.Y. Sung, I.D. Williams, B.Z. Tang, J. Phys. Chem. B 109, 17086 (2005)
Y.Q. Dong, J.W.Y. Lam, Z. Li, A. Qin, H. Tong, Y.P. Dong, X.D. Feng, B.Z. Tang, J. Inorg. Organomet. Polym. Mater. 15, 287 (2005)
B. Mi, Y.Q. Dong, Z. Li, J.W.Y. Lam, M. Haeussler, H.H.Y. Sung, H.S. Kwok, Y.P. Dong, I.D. Williams, Y. Liu, Y. Luo, Z. Shuai, D. Zhu, B.Z. Tang, Chem. Commun. 3583 (2005)
G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B.Z. Tang, D. Zhu, W. Fang, Y. Luo, J. Am. Chem. Soc. 127, 6335 (2005)
C.J. Bhongale, C.W. Chang, E.W.G. Diau, C.S. Hsu, Y.Q. Dong, B.Z. Tang, Chem. Phys. Lett. 419, 444 (2006)
Y.Q. Dong, J.W.Y. Lam, A. Qin, Z. Li, J.Z. Sun, Y.P. Dong, B.Z. Tang, J. Inorg. Organomet. Polym. Mater. 17, 673 (2007)
L. Heng, J. Zhai, A. Qin, Y. Zhang, Y. Dong, B.Z. Tang, L. Jiang, Chemphyschem 8, 1513 (2007)
Y.Q. Dong, J.W.Y. Lam, A. Qin, Z. Li, J.Z. Liu, J.Z. Sun, Y.P. Dong, B.Z. Tang, Chem. Phys. Lett. 446, 124 (2007)
L. Heng, Y.Q. Dong, J. Zhai, B.Z. Tang, L. Jiang, Langmuir 24, 2157 (2008)
Z. Li, Y.Q. Dong, J.W.Y. Lam, J.X. Sun, A.J. Qin, M. Haeussler, Y.P. Dong, H.H.Y. Sung, I.D. Williams, H.S. Kwok, B.Z. Tang, Adv. Funct. Mater. 19, 905 (2009)
X. Fan, J.L. Sun, F.Z. Wang, Z.Z. Chu, P. Wang, Y.Q. Dong, R.R. Hu, B.Z. Tang, D.C. Zou, Chem. Commun. 2989 (2008)
B.Z. Tang, X. Zhan, G. Yu, P.P.S. Lee, Y. Liu, D. Zhu, J. Mater. Chem. 11, 2874 (2001)
K. Tamao, S. Yamaguchi, M. Shiro, J. Am. Chem. Soc. 116, 11752 (1994)
A.J. Boydston, Y. Yin, B.L. Pagenkopf, J. Am. Chem. Soc. 126, 3724 (2004)
B.Z. Tang, Macromol. Chem. Phys. 209, 1303 (2008)
M. Haeussler, A.J. Qin, B.Z. Tang, Polymer 48, 6181 (2007)
M. Haeussler, B.Z. Tang, Adv. Polym. Sci. 209, 1 (2007)
B. Valeur, Molecular Fluorescence: Principle and Applications (Wiley-VCH Verlag GmbH, Weinheim, 2001)
S. Li, Q. Wang, Y. Qian, S. Wang, Y. Li, G. Yang, J. Phys. Chem. A 111, 11793 (2007)
Q. Peng, Y. Yi, Z. Shuai, J. Shao, J. Am. Chem. Soc. 129, 9333 (2007)
Q. Peng, Y. Yi, Z. Shuai, J. Shao, J. Chem. Phys. 126, 114302 (2007)
P.M. Viruela, R. Viruela, E. Orti, J.L. Bredas, J. Am. Chem. Soc. 119, 1360 (1997)
C.C. Chen, T.H. Lay, J.W. Bozzelli, J. Phys. Chem. A 107, 6451 (2003)
V. Peyrot, P. Barbier, M. Sarrazin, C. Briand, J.M. Andreu, Photochem. Photobiol. 70, 710 (1999)
X.J. Zhang, S.A. Jenekhe, Macromolecules 33, 2069 (2000)
T. Lin, Q.G. He, F.L. Bai, L.M. Dai, Thin Solid Films 363, 122 (2000)
L.S. Natrajan, A.J. Blake, C. Wilson, J.A. Weinstein, P.L. Arnold, Dalton Trans. 3748 (2004)
J.G. Muller, M. Anni, U. Scherf, J.M. Lupton, J. Feldmann, Phys. Rev. B 70, 35205 (2004)
S. Hashimoto, M. Hagiri, N. Matsubara, S. Tobita, Phys. Chem. Chem. Phys 3, 5043 (2001)
P. Blanchard, H. Brisset, A. Riou, R. Hierle, J. Roncali, J. Org. Chem. 63, 8310 (1998)
J. Jin, J. Sun, Y. Dong, H. Xu, W. Yuan, B.Z. Tang, J. Lumin. 129, 19 (2009)
G. Riess, Prog. Polym. Sci. 28, 1107 (2003)
I. Capek, Adv. Colloid Interfac. 99, 77 (2002)
I. Capek, C.S. Chern, Adv. Polym. Sci. 155, 101 (2001)
A. Khanal, Y. Inoue, M. Yada, K. Nakashima, J. Am. Chem. Soc. 129, 1534 (2007)
S. Kronholz, S. Rathgeber, S. Karthauser, H. Kohlatedt, S. Clemens, T. Schneller, Adv. Funct. Mater. 16, 2346 (2006)
H. Matsune, T. Tago, K. Shibata, K. Wakabayashi, M. Kishida, J. Nanopart. Res. 8, 1083 (2006)
M. Gradzielski, Curr. Opin. Colloid In. 9, 256 (2004)
V.P. Torchilin, Pharmaceut. Res. 24, 1 (2007)
V. Gajbhiye, P.V. Kumar, R.K. Tekade, N.K. Jain, Curr. Pharm. Design 13, 415 (2007)
N. Nishiyama, K. Kataoka, Adv. Polym. Sci. 193, 67 (2006)
K. Kostarelos, A.D. Miller, Chem. Soc. Rev. 34, 970 (2005)
K. Qi, Q.G. Ma, E.E. Remsen, C.G. Clark, K.L. Wooley, J. Am. Chem. Soc. 126, 6599 (2004)
G.S. Kwon, Crit. Rev. Ther. Drug 20, 357 (2003)
J. Jin, MS Thesis, Zhejiang University, May 2008
E.J. Fendler, J.H. Fendler, Advances in Physical Organic Chemistry, vol. 8 (Academic Press, New York, 1970)
L. Tang, J. Jin, S. Zhang, Y. Mao, J. Sun, W. Yuan, H. Zhao, H. Xu, A. Qin, B.Z. Tang, Sci. China Ser. B 52, 755 (2009)
Y. Yu, Y. Hong, C. Feng, J. Liu, J.W.Y. Lam, M. Faisal, K.M. Ng, K.Q. Luo, B.Z. Tang, Sci. China Ser. B 52, 15 (2009)
S. Kim, T.Y. Ohulchanskyy, H.E. Pudavar, R.K. Pandey, P.N. Prasad, J. Am. Chem. Soc. 129, 2669 (2007)
H.J. Tracy, J.L. Mullin, W.T. Klooster, J.A. Martin, J. Haug, S. Wallace, I. Rudloe, K. Watts, Inorg. Chem. 44, 2003 (2005)
S.J. Toal, D. Magde, W.C. Trogler, Chem. Commun. 5465 (2005)
X.J. Zhang, A.S. Shetty, S.A. Jenekhe, Macromolecules 32, 7422 (1999)
T. Maka, S.G. Romanov, M. Muller, R. Zentel, C.S. Torres, Phys. Stat. Solid. B 215, 307 (1999)
J.R. Lakowicz, J. Malicka, J. Huang, Z. Gryczynski, I. Gryczynski, Biopolymers 74, 467 (2004)
S. Setayesh, A.C. Grimsdale, T. Weil, V. Enkelmann, K. Mullen, F. Meghdadi, E.J.W. List, G. Leising, J. Am. Chem. Soc. 123, 946 (2001)
R. Iqbal, S.C. Moratti, A.B. Holmes, G. Yahioglu, L.R. Milgrom, F. Cacialli, J. Morgado, R.H. Friend, J. Mater. Sci. Mater. Electr. 97 (2000)
L. Heng, X. Wang, Y. Dong, J. Zhai, B.Z. Tang, T. Wei, L. Jiang, Chem. Asian J. 3, 1041 (2008)
K.H. Cheng, Y. Zhong, B.Y. Xie, Y.Q. Dong, Y. Hong, J.Z. Sun, B.Z. Tang, K.S. Wong, J. Phys. Chem. C 112, 17507 (2008)
C.W. Tang, S.A. VanSlyke, Appl. Phys. Lett. 51, 913 (1987)
G. Gustafsson, Y. Cao, G.M. Treacy, F. Klavetter, N. Colaneri, A.J. Heeger, Nature 357, 477 (1992)
X. Gong, D. Moses, A.J. Heeger, S. Liu, A.K.Y. Jen, Appl. Phys. Lett. 83, 183 (2003)
M.A. Baldo, D.F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M.E. Thompson, S.R. Forrest, Nature 395, 151 (1998)
M.A. Baldo, M.E. Thompson, S.R. Forrest, Nature 403, 750 (2000)
D.G. Ma, G. Wang, Y.F. Hu, Y.G. Zhang, L.X. Wang, X.B. Jing, F.S. Wang, Appl. Phys. Lett. 82, 1296 (2003)
H. Spanggaard, F.C. Krebs, Solar Energ. Mater. Solar Cells 83, 125 (2004)
P. Peumans, A. Yakimov, S.R. Forrest, J. Appl. Phys. 93, 3693 (2003)
E.W. McFarland, J. Tang, Nature 421, 616 (2003)
W.U. Huynh, J.J. Dittmer, A.P. Alivisatos, Science 295, 2425 (2002)
L. Schmidt-Mende, A. Fechtenkotter, K. Mullen, E. Moons, R.H. Friend, J.D. MacKenzie, Science 293, 1119 (2001)
H.E. Katz, A.J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.Y. Lin, A. Dodabalapur, Nature 404, 478 (2000)
J.J.M. Halls, C.A. Walsh, N.C. Greenham, E.A. Marseglia, R.H. Friend, S.C. Moratti, A.B. Holmes, Nature 376, 498 (1995)
G. Yu, J. Gao, J.C. Hummelen, F. Wudl, A.J. Heeger, Science 270, 1789 (1995)
B. Oregan, M. Gratzel, Nature 353, 737 (1991)
B.Z. Tang, H.Z. Chen, R.S. Xu, J.W.Y. Lam, K.K.L. Cheuk, H.N.C. Wong, M. Wang, Chem. Mater. 12, 213 (2000)
J.W.Y. Lam, B.Z. Tang, J. Polym. Sci. A 41, 2607 (2003)
Acknowledgements
This work was partly supported by the Research Grants Council of Hong Kong (603008, 602707, 602706, 601608), the National Basic Research Program of the Ministry of Science & Technology of China (2002CB613401), and the National Science Foundation of China (20634020). B.Z.T. thanks the support from the Cao Guangbiao Foundation of Zhejiang University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, J., Lam, J.W.Y. & Tang, B.Z. Aggregation-induced Emission of Silole Molecules and Polymers: Fundamental and Applications. J Inorg Organomet Polym 19, 249–285 (2009). https://doi.org/10.1007/s10904-009-9282-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-009-9282-8