
Abstract
In this work, we designed and synthesized a novel quinolin-based derivative which exhibited signaling behaviors for Cu2+. Upon the addition of Cu2+ to the solution of the molecule, it displayed an obvious fluorescence quenching in a linear fashion due to the formation of a 1:1 metal–ligand complex. This fluorescent sensor exhibited a rare sensitivity toward Cu(II) (the level of magnitude could be 6 × 10−8), a rapid response (<10 s) and also high selectivity toward Cu2+ over other metal ions such as Na+, K+, Ca2+, Mg2+, Ba2+, Fe3+, Mn2+, Hg2+, Pb2+, Cd2+, Cr3+, Co2+, Zn2+ and Al3+. Simultaneously, the cell imaging experiments and filter paper test demonstrated its extensive applicability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Copper, the third most abundant transition metal in human body, plays a vital role in many fundamental physiological processes. Amounts of cytosolic, mitochondrial and vesicular oxygen-processing enzymes require copper as a redox cofactor, but uncontrolled reactions of copper ions with oxygen result in the formation of reactive oxygen species (ROS), which trigger oxidative damage to proteins, nucleic acids and lipids [1, 2]. Until now, the detecting methods of Cu2+ are increasingly concerned and researched [3–5].
In recent years, there has been a growing interest in constructing optical chemosensors for fast and accurate monitoring of hazardous heavy metals. Among different kinds of chemosensors, fluorescent sensors are considerable attractive due to their high sensitivity, selectivity, rapid response and simplicity [6]. A common fluorescent sensor would involve the covalent linking of a “receptor” domain with a fluorescent fragment. The two components are intramolecularly connected together such that the binding of the target analyte causes significant influences on the photophysical properties of the fluorescent fragment, achieving the purpose of identification [7]. Quinoline-based ligands have proved popularly as fluorogenic agents for the chemical assay, whereas in recent years, quinolines and their derivatives have also been applied in copper recognition. Compared to the phenolic ligand in many excellent Cu2+ probes, as the lone electron pair of N atom at quinoline-based ligands is isolated from the electron of the aromatic ring, it is more conducive for quinoline to coordinate with metal ions [8, 9]. With the rapid development of bioanalytical technique, many fluorescent sensors have been widely utilized in biological applications for different demands, such as detecting toxic anions, cations and other compounds in living cells [10, 11]. Due to the extensive use of copper and the growing awareness of environmental protection in modern society, synthesis of fluorescent sensors for Cu2+ with good biological compatibility would have broad application prospects.
Herein we reported a new fluorescent sensor served as a Cu2+ chelator based on PET mechanism which combined quinoline with 4-(Diethylamino) salicylaldehyde. It also presented very excellent affinity for Cu2+ among other metal ions. Moreover, the living cells image experiments and filter paper test demonstrated its value in practical applications.
Experimental Section
Materials and Instruments
All the materials for synthesis were purchased from commercial suppliers and used without further purification. Solutions for spectra detection was HPLC reagent without fluorescent impurity. 1H NMR spectra were taken on a Varian mercury-400 spectrometer with TMS as an internal standard and DMSO-d 6 as solvent. HRMS spectra was analysed on an Agilent 1,290-μ TOF QII. Fluorescence spectra measurements were performed on a Hitachi F-4500 spectrofluorimeter. The pH measurements were made with Metteler-Toledo Instruments DELTE 320 pH. Cell experiment were applied on an inverted fluorescence microscope (Olympus IX-70) connected with a digital camera (Olympus, c-5050).
UV–vis and Fluorometric Analysis
The stock solution of sensor 1 was prepared in C2H5OH. Stock solutions of various cations were prepared in Tris–HCl buffer solution at pH 7.2. In titration and selectivity experiments, the test samples were prepared by placing appropriate amounts of ions stock into corresponding solution of Sensor 1. For fluorescence measurements, excitation was provided at 399 nm, and emission was collected from 420 to 600 nm, both the excitation and emission slit widths were 5 and 5 nm, respectively.
Synthesis
Preparation of Intermediate 2
A mixture of (2.0 g, 13.78 mmol) 8–hydroxyquinoline and (3.8 g, 27.56 mmol) K2CO3 was in 50 mL acetonitrile with stirring for 30 min, then (2.5 g, 15.15 mmol) ethyl bromoacetate was added. The mixture was kept stirring for 6 h at room temperature and then extracted with CH2Cl2 and H2O for 3 times, organic layer was dried on MgSO4 for 12 h before distilling the solvent. The crude product compond 3 was purified by column chromatography (silica gel, EtOAc: petroleum ether = 1:2,), and obtained as red oil with a yield of 80 % [12]. (0.5 g, 2.16 mmol) compond 3 in 5 mL MeOH was added dropwise into 1 mL hydrazine and stired for 1 h, filtered the generated precipitates and washed them with H2O and CH2Cl2 for 3 times respectively, purified the residue through silica gel column chromatography and used ethanol as eluent. The product Intermediate 2 was collected as white solid with a yield of 69 % (Scheme 1). 1H NMR (300 M DMSO, 25 °C, TMS): δ 4.40 (s, 2H), 4.76 (s, 2H), 7.26 (d, J = 6.0 Hz, 1H), 7.53 (d, J = 3.0 Hz, 2H), 7.59 (s, 1H), 8.36 (d, J = 5.3 Hz, 3H), 8.91 (d, J = 1.6 Hz, 1H). 13C NMR (300 M DMSO, 25 °C, TMS): δ 68.77, 112.19, 121.42, 122.48, 127.23, 129.6, 136.51, 140.33, 149.88, 154.42, 167.32. HRMS. Calc. for: C11H11N3O2 m/z =217.09, found [(M + 1)+] m/z = 218.1

Synthesis of Sensor 1
Synthesis of Sensor 1
To 20 mL of anhydrous DMF containing intermediate 2 (0.22 g, 1.0 mmol) was added 4-(Diethylamino) salicylaldehyde (1.93 g, 1.0 mmol) and the mixture was vigorously at 155 °C for 4 h. The reaction progress was monitored by thin-layer chromatography. After completion of the reaction, the solvent was removed and the residue was purified by column chromatography with dichloromethane/methanol (2:1 volume ratio), 0.92 g of Sensor 1 was afford as yellow solid with a yield of 43 %. 1H NMR (300 M DMSO, 25 °C, TMS): δ 1.08 (m, J = 6.0, 5.8, 6.0, 5.9 Hz, 6H), 3.45 (d, J = 6.7Hz, 4H), 4.91 (s, 2H), 5.30 (s, 1H), 6.11 (s, 1H), 6.35 (m, J = 5.7, 3.0Hz, 1H), 7.23 (d, J = 8.7 Hz,1H), 7.55 (m, J = 2.7, 9.2 Hz, 2H), 7.62 (d, J = 3.4 Hz, 2H), 8.21 (d, J = 9.4 Hz, 2H), 8.34 (s, 1H), 8.98 (s, 1H). HRMS. Calc. for: C22H24N4O3 m/z =392.45, found [(M + 1)+] m/z = 393.2
Results and Discussion
The structures of Sensor 1 were confirmed by 1H NMR and HRMS. The binding mode between Sensor 1 and Cu2+ was investigated by fluorescence spectra, NMR titration, Benesi-Hildebrand expression and Job’s plot. The spectrum titration experiments of Sensor 1 were investigated in aqueous and C2H5OH media.
Cu2+-Titration and Spectral Responses
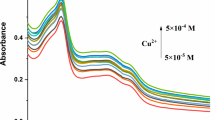
In order to gain an insight into the signaling properties of Sensor 1 toward Cu2+, titration experiments were conducted under the condition of 1 nmol/L Tris–HCl buffer solution at pH = 7.2, [V(C2H5OH)/V(H2O) = 5/5]. Sensor 1 exhibited an absorption maximum at 351 nm as illustrated in Fig. 1 ([Cu2+] = 0.1-1 μM/L), upon the addition of Cu2+, the absorption band at 351 nm decreased and had a redshift to 399 nm until the first stoichiometry. Besides, the Job’s plot (Fig. 2) exhibited the maximum absorbance appeared at w = 50 % of Sensor 1, which demonstrated that Sensor 1 and Cu2+ formed a 1: 1 stoichiometry complex. According to the linear Benesi-Hildebrand (Fig. 3) expression [13–15], the measured absorbance [1/(A0 – A)] at 399 nm varied with 1/[Cu2+] in a linear relationship (R = 0.99829). Therefore, on the basis of 1:1 stoichiometry and UV–vis titration data, the association constant of complex of Sensor 1 with Cu2+ ion was found to be 4.7735 × 108 mol−1. In the fluorescence emission, as illustrated in Fig. 4, Sensor 1(1 μM/L) alone exhibited strong fluorescence at 514 nm in water and C2H5OH (5:5) with the excitation at 399 nm. Upon the increasing addition of Cu2+ from 0 to 1 × 10−6 mol/L, the fluorescence intensity at 514 nm gradually weakened until quenching, the “turn off” sensing process could be observed under the irradiation of ultraviolet lamp (Scheme 2). Moreover, the reaction responsible for these changes reached completion well within the time frame (<10 s). Significantly, the fluorescence quenching of Sensor 1 depend on the concentration of Cu2+ was in a linear manner (R = 0.99592, Fig. 5), it indicated that Sensor 1 had potential application for quantitative determination of Cu2+. In addition, the detecting limit was calculated to be 6.6623 × 10−8 mol/L from this linear relation (based on DL = KSb1/S, Fig. 5), and this 10−8 mol/L degree detecting limit was very seldom reported in the literatures. Simultaneously, it was much lower than the TLV (10 ppb) set by the EPA.

UV–vis absorption response of Sensor 1 (1 μM/L) upon addition of different concentrations of Cu2+ (0.1 μM/L) in Tris–HCl solution [V(C2H5OH)/V(H2O) = 5:5, pH = 7.2]

Job’s plot for determining the stoichiometry of Sensor 1 and Cu2+ in Tris–HCl solution [V(C2H5OH)/V(H2O) = 5:5, pH = 7.2] [Cu2+] + [Sensor 1], the total concentration of Sensor 1 and Cu2+ was 1 μM/L

Benesi Hilderbrand plot of Sensor 1 with Cu2+

Fluorescence spectra of Sensor 1 (1 μM/L) upon addition of different concentrations of Cu2+ (0.1 μM/L) in Tris–HCl solution [V(C2H5OH)/V(H2O) = 5:5, pH = 7.2] (λex = 399 nm)

The binding mode of Sensor 1-Cu2+ system

Normalized response of the fluorescence signal to changing Cu2+ concentrations
For rationalizing the observed fluorescence quenching, two factors may be considered. First, the strong fluorescence intensity of Sensor 1 in the absence of Cu2+ ion may be attributed to radiational channel from the ππ* state of the emission of the quinoline group in the excited state, the excited electrons directly transit to the ground state to emit fluorescence. When Cu2+ ion coordinates with lone pair of acceptor moiety, the high energy level electrons will transfer to the vacated orbit of fluorophore in the excited state, and block the electrons transit to the ground state with fluorescence emission (PET), this phenomenon may cause a substantial decrease of the fluorescence intensity. Secondary, the binding between Sensor 1 and Cu2+ induces its conformation restriction, which also result in the quenched fluorescence intensity.
To further verify the configuration of Sensor 1-Cu2+, we carried out 1H NMR titration experiments in DMSO-d 6. As shown in Fig. 6, upon addition the Cu2+ dissolved in D2O, considerable changes took place in the chemical shift of protons in Sensor 1 especially in the acceptor moiety. In more detail, the dramatic change between δ 5.30 and δ 5.39 in the spectrum demonstrated the existence of proton Ha (hydroxy proton) during Cu2+ binding. Likewise, the change from δ 8.24 to δ 8.33 proved that the proton Hi (amide proton) was involved in the Cu2+ binding process. The proton Hc displayed the downshift from δ 4.91 to δ 4.99 and the chemical shift change of proton in the quinoline ring evidenced the effect of O (closed to quinoline ring) and N (in quinoline ring) in the complexing. Simultaneously, the other protons showed negligible downshift, these data further confirm the conclusion that quinoline and salicylaldehyde moiety are both involved in the coordination with Cu2+ as envisaged.

Partial 1H NMR (300 MHz) spectral changes of Sensor 1 (10 μmol/L) in DMSO-d 6 upon addition of Cu2+
Response of Sensor 1 to Various Metal Ions and Metal-ion Competition Experiments
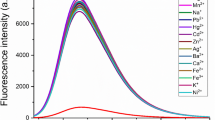
The selectivity is one of the essential requirements for a chemosensor to signal a specific species in a complex system. In selectivity experiments the fluorometric behavior of Sensor 1 (1.0 × 10−6 mol L−1) was investigated upon addition of several metal ions such as Na+, K+, Ca2+, Mg2+, Cu2+, Ba2+, Fe3+, Mn2+, Pb2+, Hg2+, Zn2+, Al3+, Cd2+, Cr3+, Co2+ (1.0 × 10−6 mol L−1) in C2H5OH–Tris buffer solution [V(C2H5OH)/V(H2O) = 5:5, pH 7.2]. Sensor 1 showed a strong flourescence emission positioned at 514 nm, upon excitation at 399 nm. As shown in Fig. 7 (black bar), only the addition of Cu2+ to Sensor 1 caused a remarkable flourescence quenching in the emission spectra, the introduction of other metal ions of Na+, K+, Ca2+, Mg2+, Cr3+, Co2+, Cd2+, Ba2+, Pb2+, Al3+, Zn2+, Hg2+ and Mn2+ slightly affected the fluorescence, whereas the metal ion like Fe3+ slightly weaken the fluorescence. But these ions would not induce any obvious interference for detecting Cu2+ in the flourescence change at 514 nm, which was probably due to several combined influences cooperating to achieve the unique selectivity for Cu2+.

Fluorescence intensities of sensor 1 (1 μM/L in the presence of various metal ions (black bar) and competition experiment (red bar) in Tris–HCl solution [V(C2H5OH)/V(H2O) = 5:5, pH = 7.2] (λex = 399 nm, λem = 514 nm)
The competition experiments were also performed for further study of the interference from other cations in the determination of Cu2+, it was conducted with addition of 1.5 equiv. Cu2+ in HQ solution to induced fluorescence quenching before mixed 10 equiv. interferential metal ions, as shown in red bars in Fig. 7, all of the fluorescence intensity exhibited still quenching. So the experimental results indicated that these ions showed no obvious interference to Cu2+ detection. Thus the fluorometric analysis above had proven that Sensor 1 could serve as an outstanding sensitive and selective fluorescent sensor for Cu2+ in our prospective.
We selected the filter paper as the supporter of the Sensor 1 to make a dipstick for the Cu2+ detecting. After dropping a solution of Sensor 1 on the neutral filter paper and drying it, a fluorescent dipstick was formed. When exposed the modified filter-paper to the aqueous solution of various metal ions under UV lamp, only Cu2+ induced the fluorescence quenching as shown in Fig. 8.

fluorescence changes of filter paper containing Sensor 1 treated with various metal ions under UV lamp
Detection of Cu2+ in Living Cells
To explore the potential biological application of this sensor, we researched the capability of Sensor 1 to track Cu2+ in live Hela (cervix carcinoma cell) cell lines. The images were obtained upon irradiation at 405 nm with a band path from 395 to 425 nm under identical exposure conditions. The living cells were first incubated with 10 μmol/L Sensor 1 in DMF for 30 min at 37 °C in 5 % CO2 atm, then washed the cells with phosphate buffered saline (PBS, pH = 7.4) 3 times, which was sufficient for the intracellular accumulation of Sensor 1 judged from its strong self-fluorescence inside living cells (Fig.9a). However, as shown in Fig.9b, intracellular fluorescence quenched after adding 20 μmol/L Cu2+ in to the cells which supplemented with Sensor 1 under the same loading conditions. So Fig. 9 displays the fluorescence images for the Hela cells stained with the Sensor 1 before and after treated with Cu2+, revealing that it can be a valuable molecular sensor for studying biological processes involving Cu2+ within living cells.

Fluorescence microscope imaging of Hela cells stained with 10 μmol/L of Sensor 1 before (a) and after (b) treating with 10 μmol/L of Cu2+. Bright-field transmission image of Hela cells incubated with Sensor 1 (1 ppm) are shown in (c)
Conclusion
We have developed a quinoline based fluorescent chemosensor for the detection of Cu2+. It displayed excellent selectivity and sensitivity to Cu2+ with a 1:1 binding mode. Upon addition of Cu2+, a striking fluorescence quenching was also detected. Moreover, the cell-permeable experiments indicated this sensor could indeed visualize the change of intracellular Cu2+ in living cells. We expect that such Cu2+ sensor will exploit potential application in biomedical and environmental detection in the future.
References
Kaur N, Singh P, Kaur S (2007) Tetrahedron 3:11724–11732
Liu JM, Wang HF, Yan XP (2011) Analyst 136:3904–3910
Wang W, Wang XL, Yang QB, Fei XL, Sun MD, Song Y (2013) Chem Commun 49:4833–4835
Karr JW, Szalai VA (2007) J Am Chem Soc 129:3796–3797
Li J, Uversky VN, Fink AL (2001) Biochemistry 40:11604–11613
Bell TW, Hext NM (2004) Chem Soc Rev 33:589–598
Wang HX, Yang L, Zhang WB, Zhou Y, Zhao B, Li XY (2012) Inorg Chima Acta 381:111–116
Tewari BB (2010) J Chem Eng Data 55:1779–1783
Ma LJ, Cao WG, Liu JL, Deng DY, Wu YQ, Yan YH, Yang LT (2012) Sensors Actuators B 169:243–247
Wang W, Wen Q, Zhang Y, Fei XL, Li YX, Yang QB, Xu XY (2013) Dalton Trans 42:1827–1833
Zhu H, Fan JL, Lu J, Hu MM, Cao JF, Wang J, Li HL, Liu XJ, Peng XJ (2012) Talanta 93:55–61
Goswami S, Aich K, Das AK, Manna A, Das S (2013) RSC Adv 3:2412–2416
Benesi HA, Hildebrand JH (1949) J Am Chem Soc 71:2703–2707
Barra M, Bohne C, Scaiano JC (1990) J Am Chem Soc 112:8075–8579
Zhu M, Yuan MJ, Liu XF, Xu JL, Lv J, Huang CS, Liu HB, Li YL, Wang S, Zhu DB (2008) Org Lett 10:1481–1484
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 385 kb)
Rights and permissions
About this article

Cite this article
Zhou, C., Song, Y., Xiao, N. et al. A Novel Highly Sensitive and Selective Fluorescent Sensor for Imaging Copper (II) in Living Cells. J Fluoresc 24, 1331–1336 (2014). https://doi.org/10.1007/s10895-014-1419-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-014-1419-7