
Abstract
In this study, the binding mode of nobiletin (NOB) with pepsin was investigated by spectroscopic and molecular docking methods. NOB can interact with pepsin to form a NOB-pepsin complex. The binding constant, number of binding sites and thermodynamic parameters were measured, which indicated that NOB could spontaneously bind with pepsin through hydrophobic and electrostatic forces with one binding site. Molecular docking results revealed that NOB bound into the pepsin cavity. Synchronous and three-dimensional fluorescence spectra results provide data concerning conformational and some micro-environmental changes of pepsin. Furthermore, the binding of NOB can inhibit pepsin activity in vitro. The present study provides direct evidence at a molecular level to show that NOB could induce changes in the enzyme pepsin structure and function.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As a digestive protease, pepsin is responsible for the most of digestion activities in the stomach [1]. It is very probable for drug to interact with pepsin when it enters the stomach. The drug may also affect the activity of pepsin and result in some adverse effects, such as hiccup singultation, nausea and vomiting. Therefore, in order to improve the safety of drug usage in clinical, it is very necessary for us to learn about the knowledge that whether the drug could interact with the pepsin, what the mechanism of this action was in this process and would such an interaction affect the activity of pepsin. Recently, in order to investigate the toxicity and metabolism mechanism of chemical components that enter the human organism through food and drug, the interactions of pepsin with these components have been reported [2–4].
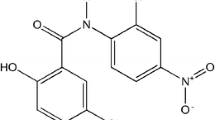
Nobiletin (NOB, 3′,4′,5,6,7,8-hexamethoxyflavone, Fig. 1), isolated from citrus fruits (such as Citrus depressa, Citrus sinensis and Citrus limon) [5, 6], has a broad spectrum of health-promoting properties including anticancer [7], antimetastatic [8], anti-inflammatory [9], antidiabetic [10], and neurotrophic activities [11]. In consideration of the possible pharmaceutical benefit, it leads us to investigate the interaction of NOB with protein, as it can provide a molecular basis for elucidating the mechanism of the drug acting and predicating unfavorable drug interaction. In recent years, several public and scientific interests have been focused on the interactions of NOB with some proteins, including human serum albumin (HSA) [12] and bovine serum albumin (BSA) [13]. However, to the best of our knowledge, little concern was placed on the binding of NOB to pepsin and its effect on the activity of pepsin. Thus, the purpose of this study was to understand the interaction mechanism of NOB with pepsin by investigating the binding parameters of the interaction and the effect of NOB on the conformation of pepsin by using multiple spectroscopic techniques and molecular modeling. Moreover, the effect of NOB on the pepsin activity in vitro was also investigated. This study provides basic data for clarifying the binding mechanism of NOB with pepsin and is help for understanding the symptoms of indigestion after oral administration.

Molecular structure of nobiletin (NOB)
Experimental
Reagents
The porcine pepsin was purchased from Sigma-Aldrich Chemical Co. (USA) and nobiletin (NOB) was obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). All the chemicals were of analytical-reagent grade and used without further purification. NOB was dissolved in methanol to form a 1.0 × 10−3 mol L−1 solution, which was used to determine the binding sites of NOB on pepsin. Citric acid-sodium citrate buffer solutions (0.2 mol L−1) containing 0.1 mol L−1 NaCI were prepared to adjust the acidity of the system pH = 2.0, which is the most common pH for pepsin digests. Water was purified with a Milli-Q purification system (Barnstead, USA).
Equipment and Spectral Measurements
The fluorescence spectra were recorded on Hitachi F-2500 spectrofluorimeter equipped with 1.0 cm quartz cells. The excitation wavelength was 280 nm for all cases with an excitation and emission band pass (slit) of 10 nm. The experimental temperature was maintained by recycling water throughout the quartz cell. The UV–vis spectrum was recorded at room temperature on a Shimadzu UV-2450 spectrophotometer (Shimadzu, Japan) equipped with 1.0 cm quartz cells.
Procedures
The fluorescence measurements were carried out by successive addition of the solution of NOB to a fixed amount of pepsin (to give a final concentration of 2.5 × 10−5 mol L−1) in each tube. The final volume was made up to 5.0 mL with citric acid-sodium citrate buffer (pH = 2.0). Thus, a series of solutions containing different amount NOB and a definite amount of pepsin were obtained. The fluorescence spectra were then measured (excitation at 280 nm and emission wavelengths of 290–450 nm) at 288 and 310 K, respectively. All solutions were mixed thoroughly and kept 20 min before measurement.
The synchronous fluorescence spectra of pepsin in the presence of NOB were recorded at 288 K and the D-value (∆λ) between excitation wavelength and emission wavelength were stabilized at 15 nm or 60 nm. The three-dimensional fluorescence spectra were performed under the following conditions: the emission wavelength range was selected from 270 to 500 nm, the initial excitation wavelength was set to 200 nm, and the scanning number was 15 with the increment of 10 nm. The UV–vis absorbance spectra of NOB-pepsin system were recorded at 288 K.
Molecular Docking Investigation
Docking calculations were carried out using AutoDock 4.0. The structure of NOB was generated by Chem 3D Ultra and optimized by density functional theory at B3lyp/6 − 31 + g (d) level implemented in Gaussian 03 until all egienvalue of the Hesssian matrix were positive. With the aid of AutoDock 4.0, the ligand root of NOB was detected and rotatable bonds were defined.
The crystal structure of pepsin (protein ID: 5PEP) was downloaded from Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). All water molecules were removed and the polar hydrogen and the Gasteiger charges were added at the beginning of docking study. To recognize the binding sites in pepsin, docking was carried out with setting of grid box size 90 Å × 100 Å × 90 Å along x, y, z axes covering whole protein with Kollman charges. The grid center was set at −17.802 Å, 40.376 Å and 86.848 Å. At first, AutoGrid was run to generate the grid map of various atoms of the ligand and receptor. After the completion of grid map, ligand flexible docking simulations were performed with 100 runs and 2,500,000 energy evaluations, 25000 numbers of generations, 50 GA populations and root mean square cluster tolerance 2.0 Å per run. Finally, the lowest energy conformation was used for docking analysis.
Pepsin Activity Measurement
The enzyme activity was detected by the method detailed by Anson with some modifications [14]. Pepsin (2.5 × 10−6 mol · L−1) in citric acid-sodium citrate buffer (pH = 2.0) was mixed with various concentrations of NOB (0, 1.0, 2.0, 4.0, 6.0, 8.0, 10.0, 12.0 × 10−5 mol · L−1) at 37 °C for 10 min, and then 1.0 mL of 5 % bovine hemoglobin solution was added in. After 10 min, 5.0 mL of 5 % trichloroacetic acid was added to terminate the reaction. The mixture was put there for 10 min statically, and then centrifuged at 5000 rpm for 15 min. After addition of 3.0 mL of NaOH and 300 μL of Fehling’s solution to the supernatant, the mixture was incubated at 37 °C for 15 min and then the value of OD660 was measured using a spectrophotometer. The activity of pepsin can be calculated by the following equation:
Results and Discussion
Characterization of the Binding Interaction of NOB with Pepsin by Fluorescence Measurements
Fluorescence Quenching
Fluorescence methods have been widely used to investigate the interaction between ligands and proteins and can give information about the quenching mechanism, binding constants and binding sites [15]. We utilized the technique to study the interaction between pepsin and NOB. The fluorescence spectra of pepsin at various concentrations of NOB were shown in Fig. 2. The fluorescence intensity of pepsin decreased regularly with an increasing concentration of NOB, which indicated that NOB can bind to the pepsin and alter the structure of it.

The fluorescence emission spectra of pepsin in the presence of increasing amounts of NOB. Peak from up to down C NOB = (0, 3.0, 6.0, 9.0, 12.0, 15.0,18.0, 21.0, 24.0, 30.0) × 10−6 mol · L−1, CPepsin = 2.5 × 10−5 mol · L−1
The different mechanisms of quenching are usually classified as either dynamic quenching or static quenching, which can be distinguished by their different dependence on temperature and viscosity, or preferably by lifetime measurements. Dynamic quenching and static quenching are caused by collisional encounters and ground-state complex formation between fluorophores and quenchers, respectively. As higher temperatures result in larger diffusion coefficients, dynamic quenching constants are expected to be higher with increasing temperature. In contrast, the increase of temperature is likely to result in decreased stability of complexes, thus the values of the static quenching constants are expected to be smaller [16]. To confirm the mechanism, we used the well-known Stern-Volmer equation:
where F 0 and F are the fluorescence intensities before and after the addition of the quencher, respectively. K q is the bimolecular quenching constant,τ 0 is the lifetime of the fluorophore in the absence of the quencher (τ 0 = 10−8 s) [17], [Q] is the concentration of the quencher, and K sv is the Stern-Volmer quenching constant.
Fluorescence intensity data were analyzed according to F 0/F versus [Q] at 293 and 310 K (shown in Fig. 3). Equation (1) was applied to determine K sv and K q by a linear regression plot of F 0/F versus [Q]. The calculated K sv and K q at two different temperatures are listed in Table 1. The K sv values decreased with increasing temperature. Moreover, the maximum dynamic quenching constant K q of the various quenchers is 2.0 × 1010 L mol−1 S−1 [18]. However, the values of K q at 293 and 310 K are greater than 2.0 × 1010 L mol−1 S−1. Thus, the results indicated that the overall quenching was dominated by a static quenching mechanism, and in this process a NOB-pepsin complex was formed.

Sterm-Volmer plots for the quenching of pepsin by NOB at different temperatures (n = 3)
Binding Constant and Binding Capacity
For the static quenching interaction, when small molecules bind independently to a set of equivalent sites on a macromolecule, the equilibrium between free and bound molecules could be represented by the equation [19]:
where F 0 and F are fluorescence intensities with and without quencher, respectively. K a is static quenching constant, n is the number of binding sites per pepsin molecule, [Qd] and [Qp] are the concentration of drug molecule and protein, respectively. By plot of log(F 0 − F)/F versus log{1/[[Qd] − (F 0 − F)[Qp]/F 0]} (Fig. 4), the number binding sites n and binding constant K a of the interaction between NOB and pepsin can be calculated and the results are summarized in Table 2. The number of binding sites n is approximately equal to 1, indicating that there was one binding site in pepsin for NOB during their interaction. The value of K a is of the order of 105 L mol−1, indicating that a strong interaction exists between NOB and pepsin.

Double-log plots of NOB quenching effect on pepsin fluorescence at different temperatures (n = 3)
Binding Forces
The action forces contributing to protein interactions with small molecular substrates often include van der Waals interaction, hydrophobic force, electrostatic interaction and hydrogen bond. Thermodynamic parameters are important for confirming the non-covalent acting forces. Ross and Subramanian have summed up the thermodynamic laws to determine the types of binding with various interactions [20]. If ΔH < 0 and ΔS < 0, van der Waals and hydrogen bond interactions play the main roles in the binding reaction. If ΔH > 0 and ΔS > 0, hydrophobic interactions are dominant. If ΔH < 0 and ΔS > 0, the main force is an electrostatic force. When the temperature range is not too wide, the enthalpy change (ΔH) can be regarded as a constant. The enthalpy change, free-energy change and the entropy change for the interaction between NOB and pepsin were calculated based on the van’t Hoff equation (Eq. 3) and thermodynamic equations (Eqs. 4 and 5):
where K is the binding constant at the corresponding temperature, R is the gas constant, T is absolute temperature. The values of ΔG, ΔH and ΔS are listed in Table 3. The ΔG at 293 and 310 K are negative, indicating that the binding process is spontaneous. The negative ΔH and positive ΔS mean that electrostatic interactions play a major role in the formation of the NOB-pepsin complex. However, the value of ΔH is close to zero, indicating that hydrophobic interactions also play a very important role in the formation of the complex [16]. Therefore, the interaction force in the binding process is mainly electrostatic and hydrophobic interactions.
Energy Transfer from Pepsin to NOB
Fluorescence resonance energy transfer (FRET) is a non-destructive spectroscopic method that can monitor the proximity and relative angular orientation of fluorophores. The donor and acceptor fluorophores can be entirely separated or attached to the same macromolecule. Energy transfer can take place through direct electrodynamic interaction between the primarily excited molecule and its neighbors [21]. The ‘spectroscopic ruler’ is suitable for distance measurement over several nanometers [22]. The distance between the donor (pepsin) and the acceptor (NOB) was estimated by the following Eq. (6) according to FÖrster’s non-radiative energy transfer theory [23]:
where E denotes the efficiency of energy transfer between the donor and the acceptor, and R 0 is the critical distance when the efficiency is 50 %, which can be estimated by the following equation:
where k 2 is the orientation factor related to the geometry of the donor and acceptor of dipoles, n is the average refracted index of medium in the wavelength range where spectral overlap is significant, Φ is the fluorescence quantum yield of the donor, and J is the effect of the spectral overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor (Fig. 5), which can be calculated by the equation:

Overlapping of fluorescence spectra of pepsin (Cpepsin = 2.5 × 10−5 mol L−1) with absorption spectra of NOB (C NOB = 1.67 × 10−4 mol L−1)
where F(λ) is the fluorescence intensity of donor in wavelength λ with dimensionless, ε(λ) denotes the extinction coefficient of the acceptor at λ.
In the present case, k 2 = 2/3, n = 1.36 and Φ = 0.15 [24]. According to Eqs. (6)–(8), it can be calculated that J = 2.026 × 10−14 cm3 · moL−1 · L, E = 0.745, R 0 = 0.406 nm and r = 0.339 nm. The donor-to-acceptor distance is less than 8 nm, indicating that the energy transferring from pepsin to NOB occurs with high possibility. This was in accordance with conditions of FÖrster theory of non-radioactive energy transfer and indicated again a static quenching between NOB and pepsin.
Conformational Investigations
Synchronous Fluorescence Spectroscopy
Synchronous fluorescence spectroscopy can be used to explore the change of the molecular microenvironment in the vicinity of chromophores (such as tryptophan (Trp) and tyrosine (Tyr)) and it involves simultaneous scanning of the excitation and emission monochromators while maintaining a constant wavelength interval between them [15]. When the wavelength interval (∆λ) is stabilized at 15 nm, the synchronous fluorescence of pepsin characteristic of a Tyr residue. And when ∆λ is fixed at 60 nm, a spectrum characteristic of Trp residues is obtained [25]. As shown in Fig. 6a, the emission maximum of Tyr kept the position with the increasing concentration of NOB, which indicates that NOB has little effect on the microenvironment of Tyr residues in pepsin. However, in Fig. 6b the Trp peak was red-shifted with the increasing concentration of NOB, which indicates that the hydrophobicity of the Trp residues decreased and the Trp buried in the nonpolar hydrophobic cavities were moved to a more hydrophilic environment. Moreover, it can be seen from Fig. 7 that the slope was higher when ∆λ was 60 nm, which indicated that NOB was closer to the Trp residues than to the Tyr residues and the microenvironments of Trp residues were influenced more than those of Tyr residues.

Synchronous fluorescence spectra of interaction between pepsin and NOB at a Δλ = 15 nm and b Δλ = 60 nm at room temperature. Peak from up to down: CNOB = (0, 3.0, 6.0, 9.0, 12.0, 15.0,18.0, 21.0, 24.0, 30.0) × 10−6 mol · L−1, CPepsin = 2.5 × 10−5 mol · L−1

The quenching of pepsin synchronous fluorescence by NOB. CPepsin = 2.5 × 10−5 mol · L−1, (◆) ∆λ = 15 nm and (■) ∆λ = 60 nm (n = 3)
Three-Dimensional Fluorescence Spectra
Three-dimensional fluorescence contour spectra are a rising fluorescence analysis technique in recent years [26] and can give additional information or evidence regarding the conformational changes of pepsin in presence of NOB. Figure 8 presented the three-dimensional fluorescence contour spectrum of pepsin (a) and NOB-pepsin (b). It can be seen from Fig. 8a that the three-dimensional fluorescence contour spectra of pepsin shows contour maxims at λ ex/λ em = 280/340 nm arising by π-π* transition of aromatic amino acids in pepsin. In Fig. 8b, the pepsin fluorescence peak shifted to λ ex/λ em = 285/360 nm. The stoke shift of pepsin (λ em − λ ex = 60 nm) and NOB-pepsin (λ em − λ ex = 75 nm) was also different obviously. The above phenomena and the analysis of the fluorescence characteristic of the peaks revealed that the binding of NOB to pepsin induced some micro-environmental and conformational changes in pepsin, a complex between NOB and pepsin has formed. On the other hand, the relative fluorescence intensity of pepsin peak was 423 in the absence of NOB. After the addition of NOB, the relative fluorescence intensity of peak decreased to 173. The relative fluorescence intensity of pepsin peak decreased a lot after the addition of NOB, which implied that the peptide chain structure of pepsin was changed and this result was consistent with that of synchronous fluorescence spectra.

The three-dimensional fluorescence contour spectra of a pepsin, and b NOB–pepsin system. a CPepsin = 2.5 × 10−5 mol · L−1, CNOB = 0.0 mol · L−1; b CPepsin = 2.5 × 10−5 mol · L−1, CNOB = 3.0 × 10−5 mol · L−1
Molecular Docking
To identify the precise binding sites on pepsin, a docking program was performed to simulate the binding mode between pepsin and NOB. The lowest energy-ranked results of six NOB-pepsin conformations are listed in Table 3. The ranked binding energy of the six conformations is −5.7, −5.57, −5.33, −5.13, −4.99 and −4.84 kcal mol−1. For all the six simulated binding conformations, NOB binds into the pepsin cavity. Moreover, the exact binding sites of NOB on pepsin are different among the six conformations. The exact binding site of NOB on pepsin with the lowest binding free energy is presented in Fig. 9. This docking revealed the most likely binding site in the enzyme. As shown in Fig. 9a, NOB binds into the pepsin cavity, which locates between domainIand domainII. The amino acid residues lining this binding site are 20 amino acid residues, which includes 12 hydrophobic amino acid residues (such as Ile30, Gly34, Tyr75, Gly76, Gly78, Phe111, Leu112, Phe117, Ile120, Tyr189, Gly217 and Val292) and 8 hydrophilic amino acid residues (such as Glu13, Asp32, Ser35, Thr74, Thr77, Asp215, Thr218 and Met290) (Fig. 9b). Therefore, the essential driving forces of NOB binding to this site were mainly hydrophobic and electrostatic forces. Moreover, there also exist two hydrogen bonds between NOB and Gly34 (the distance, 2.800 Å), and Thr77 (the distance, 2.2838 Å) residues of pepsin.

Docked pose corresponding to the minimum energy conformation for NOB binding to pepsin. a The hydrophobicity of pepsin with NOB. b Detailed illustration of the amino acid residues lining the binding site in the pepsin cavity. Green molecule displays NOB; broken lines display hydrogen bonds
In addition, as shown in Fig. 3, NOB also binds with two aspartate residues (Asp32 and Asp215). It is reported that the catalytic site of pepsin was formed by these two amino acid residues for the protein to be active [27]. Thus, according to docking and synchronous fluorescence results, NOB bound directly into the enzyme cavity site and the binding of NOB into the enzyme cavity influenced the microenvironment of the catalytic site, which would affect the activity of pepsin.
Effect of NOB on Pepsin Activity
In order to reveal whether NOB can affect of the activity of pepsin after it enters the organism through food and drug, the effect of NOB on the pepsin activity in vitro was investigated. As shown in Fig. 10, with the increase of NOB concentration, the relative pepsin activities were decreased significantly and the 50 % relative activity was about 6.69 × 10−5 mol L−1. The result implies that NOB can inhibit pepsin activity.

Effect of NOB on pepsin activity in vitro (n = 3)
Conclusion
In this study, the binding of NOB with pepsin has been investigated by multi-spectroscopic and molecular docking methods. NOB effectively quenched the fluorescence of pepsin by a static quenching process. Based on the results of binding capacity, calculated thermodynamic parameters and molecular docking study, it was concluded that the NOB could spontaneously bind with pepsin mainly through hydrophobic and electrostatic forces. The synchronous and three-dimensional fluorescence spectra revealed that the microenvironment and conformation of pepsin were demonstrably changed in the presence of NOB. Since the binding of NOB affected the microenvironment of the pepsin activity site, NOB caused the inhibition of pepsin activity.
References
Gole A, Dash C, Rao M, Sastry M (2000) Encapsulation and biocatalytic activity of the enzyme pepsin in fatty lipid films by selective electrostatic interactions. Chem Commun 4:297–298
Huang YB, Yan J, Liu BZ, Yu Z, Gao XY, Tang YC, Zi YQ (2010) Investigation on interaction of prulifloxacin with pepsin: a spectroscopic analysis. Spectrochim Acta A 75:1024–1029
Zhang HM, Cao J, Fei ZH, Wang YQ (2010) Investigation on the interaction behavior between bisphenol A and pepsin by spectral and docking studies. J Mol Struct 1021:34–39
Boeris V, Micheletto Y, Lionzo M, Silveira NP, Pico G (2011) Interaction behavior between chitosan and pepsin. Carbohyd Polym 84:459–464
Chen J, Montanari AM, Widmer WW (1997) Two new polymethoxylated flavones, a class of compounds with potential anticancer activity, isolated from cold pressed Dancy tangerine peel oil solids. J Agric Food Chem 45:364–368
Nagata U, Sakamoto K, Shiratsuchi H, Ishi T, Yano M, Ohta H (2006) Flavonoid composition of fruit tissues of citrus species. Biosci Biotechonol Biochem 70:178–192
Silalahi J (2002) Anticancer and health protective properties of citrus fruit components. Asia Pac J Clin Nutr 11:79–84
Sato T, Koike L, Miyata Y, Hirata M, Mimaki Y, Sashida Y, Yano M, Ito A (2002) Inhibition of activator protein-1 binding activity and phosphatidylinositol 3-kinase pathway by nobiletin, a polymethoxy flavonoid, results in augmentation of tissue inhibitor of metalloproteinase-1 production and suppression of production of matrix metalloproteinases-1 and −9 in human fibrosarcoma HT-1080 cells. Cancer Res 62:1025–1029
Lin N, Sato T, Takayama Y, Mimaki Y, Sashida Y, Yano M (2003) Novel anti-inflammatory actions of NOB, a citrus polymethoxy flavonoid, on human synovial fibroblasts and mouse macrophage. Biochem Pharmacol 65:2065–2071
Saito T, Abe D, Sekiya K (2007) Nobiletin enhances differentiation and lipolysis of 3T3-L1 adipocytes. Biochem Biophys Res Commum 357:371–376
Matsuzaki K, Miyazaki K, Sakai S, Mimaki Y, Yamakuni T, Ohizumi Y (2008) Nobiletin, a citrus flavonoid with neurotrophic action, augments protein kinase A-mediated phosphorylation of the AMPA receptor subunit, GluR1, and the postsynaptic receptor response to glutamate in murine hippocampus. Eur J Pharmacol 578:194–200
Yue YY, Zhang YH, Li Y, Zhu JH, Qin J, Chen XG (2008) Interaction of nobiletin with human serum albumin studied using optical spectroscopy and molecular modeling methods. J Lumin 128:513–520
Yang ZX, Wen YQ, Zhang YY, Yao SZ (2009) Studies on the interaction of nobiletin and bovine serum albumin by spectral methods. J Anal Sci 25:527–532
Anson ML (1938) The estimation of pepsin, trypsin, papain, and the cathepsin with haemoglobin. J Gen Physiol 22:79–89
Teng Y, Zhang H, Liu RT (2011) Molecular interaction between 4-aminoantipyrine and catalase reveals a potentially toxic mechanism of the drug. Mol Biosyst 7:3157–3163
Zhang YZ, Zhou B, Zhang XP, Huang P, Li CH, Liu Y (2009) Interaction of malachite green with bovine serum albumin: determination of the binding mechanism and binding site by spectroscopic methods. J Hazard Mater 163:1345–1352
Lakowicz JR, Weber G (1973) Quenching of protein fluorescence by oxygen. Detection of structural fluctuations in proteins on the nanosecond time scale. Biochemistry 12:4171–4179
Chi ZX, Liu RT, Zhang H (2010) Noncovalent Interaction of Oxytetracycline with the Enzyme Trypsin. Biomacromolecules 11:2454–2459
Bi SY, Ding L, Tian Y, Song DQ, Zhou X, Liu X, Zhang HQ (2004) Investigation of the interaction between flavonoids and human serum albumin. J Mol Struct 703:37–45
Ross PD, Subramanian S (1981) Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry 20:3096–3102
Hu YJ, Liu Y, Hou AX, Zhao RM, Qu XS, Qu SS (2004) Studies on the interaction between rare-earth salts of heteropoly EuHSiMoW2O40 center dot 25H(2)O and bovine serum albumin. Acta Chim Sin 62:1519–1523
Yang MM, Yang P, Zhang LW (1994) Study on interaction of caffeic acid series medicine and alburmin by fluorescence method. Chin Sci Bull 39:734–739
Förster T (1948) Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann Phys 437:55–75
Jin J, Zhang X (2008) Spectrophotometric studies on the interaction between pazufloxacin mesilate and human serum albumin or lysozyme. J Lumin 128:81–86
Zhu JF, Zhang X, Li DJ, Jin J (2007) Probing the binding of flavonoids to catalase by molecular spectroscopy. J Mol Struct 843:38–44
Dong CY, Ma SY, Liu Y (2013) Studies of the interaction between demeclocycline and human serum albumin by multi-spectroscopic and molecular docking methods. Spectrochim Acta A 103:179–186
Antonov VK, Ginodman LM, Kapitannikov YV, Barshevskaya TN, Gurova AG, Rumsh LD (1978) Mechanism of pepsin catalysis: general base catalysis by the active-site carboxylate ion. FEBS Lett 88:87–90
Acknowledgement
We gratefully acknowledge the financial support of the National Natural Science Foundation of China (U1304823) and the Natural Science Project of the Education Department of Henan Province (201212459).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zeng, Hj., Qi, T., Yang, R. et al. Spectroscopy and Molecular Docking Study on the Interaction Behavior Between Nobiletin and Pepsin. J Fluoresc 24, 1031–1040 (2014). https://doi.org/10.1007/s10895-014-1379-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-014-1379-y