
Abstract
The present work aims to investigates the native fluorescence and time resolved fluorescence spectroscopic characterization of oral tissues under UV excitation. The fluorescence emission spectra of oral tissues at 280 nm excitation were obtained. From the spectra, it was observed that the alteration in the biochemical and morphological changes present in tissues. Subsequently, the Full width at Half Maximum (FWHM) of every individual spectra of 20 normal and 40 malignant subjects were calculated. The student’s t-test analysis reveals that the data were statistically significant (p = 0.001). The fluorescence excitation spectra at 350 nm emission of malignant tissues confirms the alteration in protein fluorescence with respect to normal counterpart. To quantify the observed spectral differences, the two ratio variables R1 = I275/I310 and R2 = I310/I328 were introduced in the excitation spectra. Among them, the Linear Discriminant Analysis (LDA) of R1 reveals better classification with 86.4 % specificity and 82.5 % sensitivity. The fluorescence decay kinetics of oral tissues was obtained at 350 nm emission and it was found that the decay kinetics was triple exponential. Then the ROC analysis of fractional amplitudes and component lifetime reveals that the average lifetime shows 77 % sensitivity and 70 % specificity with the cut off value 4.85 ns. Briefly, the average lifetime exhibits better statistical significance when compared to fractional amplitudes and component lifetimes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cancer is a dreaded disease, having continuous increase in the mortality and morbidity rate in worldwide. In addition to the 20 million existing cancer cases, ten million new cases are being identified each year. It was reported that the six million deaths are due to cancer, of which 43 % are due to consumption of tobacco, alcohol, irregular lifestyle, poor food habits and less care of health condition [1]. Of all the cancers, oral cancer is the most dominant cancer in developing countries like India and other south Asian countries. A study reveals that, oral cancer accounts for 22.9 % cancer deaths in India and the primary etiology being the usage of tobacco either smoked or chewed [2]. In spite of various therapeutic advancements, still the improvement in the survival rate not up to the expectation.
Oral cancer commonly affects the outer layer of the oral cavity (palate, lip, mucosa, etc.,) which are generally preceded by the dysplastic conditions such as erythroplakia and leukoplakia [3]. As these preinvasive lesions have the probability of transforming into malignancy, early detection of the same is mandatory for the effective treatment. Further, oral cavity is highly complex, as its tissues are heterogeneous with respect to its site and hence their histology, metabolism and keratin content are differing [3]. Hence, it may be difficult to discriminate different pathological conditions of the oral cavity using visual inspection and subsequent histological examination [3]. By considering various limitations of conventional biopsy techniques, many have realized the need for the development of a non invasive and objective diagnostic technique to improve the existing diagnostic potential and thereby improve the quality of life and survival rate of the patients [3–5].
Among various optical spectroscopic techniques in particular, fluorescence spectroscopy have been established as the promising diagnostic tool in cancer diagnosis to probe the biochemical and morphological conditions of normal and diseased state of tissues [4]. Tissue consists of several endogeneous fluorophores such as tryptophan, tyrosine, phenylalanine, and Nicotinamaide Adenine Dinucleotide of reduced form (NADH), Flavin-Adenine Dinucleotide (FAD), collagen [5–7]. Fluorescence associated with these molecules is reflective of their concentration and distribution as well as the photophysicochemical properties of their environment [5]. Of all the endogenous fluorophores, tryptophan is the predominant emitting fluorophore in tissues under UV excitation than tyrosine [8]. Tryptophan is the essential amino acid in human metabolism and the alteration of this amino acid level can be used to probe the diseased state of cells and tissues. It is also known to be the popular fluorescent probe which there by access different conformational state and structural heterogeneity of the protein [9–12].
Photophysical properties of various intrinsic biomolecules and its structural alteration is found to be a useful parameter to extract the additional information about their functional state, morphology and its local microenvironmental alteration in tissues [13]. Thus, the photophysical characterization of tryptophan is necessary to understand the internal dynamics of protein under malignant transformation of oral tissues and it can be used to study the conformational and microenvironmental changes in cells and tissues. Many have reported on the diagnosis of various neoplastic conditions by probing endogenous fluorophores in tissues of different anatomical sites viz. G.I.tract [14, 15], breast [16, 17], cervix [13, 18], stomach [19] and oral cavity [4, 20, 21] using steady state fluorescence spectroscopy [22]. In addition to the conventional steady state fluorescence spectroscopy, other complementary techniques have also been reported [21, 23]. Though fluorescence spectroscopy is well established technique, various limitations such as dependent on lamp intensity, concentration of fluorophores and photo bleaching tend to look for alternate modality to overcome this limitation.
In the last decade, time resolved fluorescence technique [22] has been emerged as an effective tool in cancer diagnosis due to its unique characteristics such as independent of sample concentration, lamp intensity and to study overlapping fluorescence. Further, advantages like its high sensitivity to local microenvironment (temperature, pH, viscosity and polarity) and ability to study fluorophores having same emission wavelength with different fluorescence decay makes this technique more attractive [24]. Under these circumstances, it warrants to note that protein fluorescence is highly sensitive to tissue structure and cellular metabolism. This is because, fluorescence of tryptophan in tissues is highly sensitivity to local environment and their different conformation states of protein [9, 10]. Alternatively, lifetime spectroscopy in cancer diagnosis recently gained more interest to probe the native fluorophores in its excited state kinetics.
For instance, Swaminathan et al. [10] have extensively studied the photophysical properties of single as well as multi tryptophan both in its native and random coil state using fluorescence lifetime spectroscopy. In addition to that, Pan et al. [11] investigated how the polarity of the indole ring structure affects the native fluorescence as well as the fluorescence decay kinetics of the same. Very recently, Albani et al. [12, 25] investigated extensively on the origin of tryptophan fluorescence lifetimes in water as well as in proteins. They showed that the third fluorescence lifetime component arises depending upon the solvent when tryptophan interacts with the hydrophilic or hydrophobic chemical structure present in water. Subsequently, it was also reported that the tryptophan fluorescence decay in proteins can only be explained with triple exponential lifetime.
Based on this, Tata et al. [26] investigated the steady state and excited state kinetics of normal and cancerous in vitro rat kidney tissues using mode-locked argon ion pulsed laser system at 353 nm in the wavelength region of 500–550 nm and 550–600 nm which reveals changes in lifetimes with respect to changes in the local microenvironment of the tissues. The decay kinetics of benign and malignant breast tissues at 310 nm excitation was investigated by Pradhan et al. [27]. From the study, it reveals that the decay kinetics was fitted with bi-exponential and hence the two lifetimes obtained (slow and fast components) discriminates the malignant from benign tissues.
Subsequently, the study was extended to in vitro studies on metastatic and non-metastatic cell lines from different species were investigated under UV (310 nm) and visible excitation (350 nm) wavelengths. The decay kinetics at 350 and 450 nm were due to tryptophan and NADH respectively [8]. An in vivo study on fluorescence lifetime spectroscopy of normal and pre malignant human oral tissues at 405 nm excitation was observed and subsequent statistical analysis of the same reveals that the good discrimination between normal and premalignant oral tissues is observed [28]. Recently, Meier et al. [29] investigated the time-resolved laser-induced fluorescence in head and neck carcinoma using 337 nm pulsed laser source as excitation. It was observed that at 390 nm, it shows better discrimination than other wavelengths. Similarly, many reported on various kinds of tissues such as cortical sarcoma [30] heart [31], lung [32], and skin [24] tissues either in vivo or in vitro conditions. To the best of our knowledge, limited number of studies has been reported on decay kinetics of oral tissues. Further, to interpret the internal dynamics of protein in tissues under different microenvironment, it is essential to probe the photophysiochemical properties using native fluorescence and time resolved fluorescence spectroscopy.
In this context, the present work aims to investigate the native fluorescence and time resolved fluorescence spectroscopy of normal and malignant oral tissues at 350 nm emission to findout whether this modality exhibits any diagnostic potential or not. In addition to this, attempts have also been made with the help of statistical analysis of raw data using SPSS 19.0, to discriminate malignant from normal tissues.
Materials and Methods
Tissue Sample
In this study 22 normal and 40 malignant oral tissues of different pathological conditions were obtained from patients attending the Government Arignar Anna Cancer Hospital, Kancheepuram, Chennai, India. The consent forms were properly obtained from the patients and hospital review board was approved to obtain the samples from the corresponding hospital. Immediately after resection the tissue samples were rinsed with 0.9 % physiological saline (pH = 7.4) and stored at 4 °C in an ice box until the fluorescence and time resolved fluorescence experiments which took place within 4–5 h of resection. At the time of measurements, the sample was thawed and was kept moistened with saline. The tissue sample was placed in a sample holder and their corresponding measurements were performed [33].
Fluorescence Spectroscopic Characterization
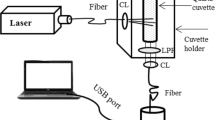
The native fluorescence spectroscopic characterization of tissues was carried out using spectroflurometer of model Fluoromax-2, SPEX, Edison, New Jersey, USA. The excitation source, 150 W Ozone free Xenon arc lamp, coupled to the monochromator, delivers light to the sample at a desired wavelength and the fluorescence emission from the sample were collected by an emission monochromator connected to a photomultiplier tube (R928P; Hamamatzu, Shizuoka-Ken, Japan). The gratings on the excitation and emission monochromators had a groove density of 1,200 grooves/mm−1 and were blazed at 330 nm and 500 nm, respectively. Excitation and emission slit width were fixed as 5 nm. The acquisition interval and the integration time were maintained as 1 nm and 0.1 s respectively [6, 7].
Fluorescence Lifetime Measurements
Lifetime measurements were made using Time Correlated Single Photon Counting (TCSPC) System on Fluorolog-3 (HORIBA Jobin Yvon, INC, Edison, NJ) by exciting the sample using 280 nm Nano LED (Pulse Width: <1 ns), with a fast response red sensitive PMT(R928P, Hamamatsu Photonics, Shizuoka-Ken , Japan) detector. The fluorescence emission was collected at 90° angle from the path of the excitation light source. The electrical signal was amplified by a pulse amplifier (Model No. TB-02, Horiba) fed to the single channel constant fraction timing discriminator (Model No. 6915, Philips Scientific, Mahawah, NJ). The first detected photon was used as a start signal by a time-to-amplitude converter (TAC), and the excitation pulse triggered the stop signal. The multichannel analyzer (MCA) recorded the repetitive start-stop signals from the TAC and generated a histogram of photons as a function of time-calibrated channels (55.7 ps/channel) until the peak signal reached 10,000 counts for 350 nm emission. The instrument response function was obtained using the Rayleigh scatter of Ludox-40 (40 weight percentage suspension in water; Sigma-Aldrich) at 280 nm excitation. Decay analysis software (DAS6 v6.0, Horiba) was used to extract the lifetime components. The goodness of fit was judged by chi-square values, Durbin-Watson parameters, as well as visual observations of fitted line, residuals, and autocorrelation functions [34]. The average lifetime (τ avg) with amplitude (α i) and lifetime τi, i =1, 2, 3 were calculated using the following Equation.
Statistical Analysis
Introducing Ratio Variables
To quantify the results and estimate the diagnostic potentiality of the present technique, two ratio variables were introduced. These ratio parameters were computed using fluorescence intensities at different excitation wavelengths, whose significant variation in the spectral signatures were observed [6].
Linear Discriminant Analysis
The Linear Discriminant Analysis (LDA) were also performed as described by our earlier reports [6, 7] for two ratio variables (R1 = I275/I310 and R2 = I310/I328). The analysis was performed across two groups, tissues from 22 normal subjects and 40 oral malignancy subjects were histopathologically confirmed as cancer patients. The LDA was performed using SPSS-19.0. The discriminant analysis used a partial F-test (F to enter 3.84; F to remove 2.71). The classification function of one group could discriminate another group in the analysis. To check the reliability of our analysis, leave one-out cross-validation (LOOCV) was used. In this procedure, discriminant scores of one particular case were eliminated and discriminant analysis was used in the analysis. The resulting algorithm was then used to classify the excluded case. This process known as leave-one-out cross validation or jack knife cross-validation provided optimal use of a small data set to validate the performance of a decision surface without bias. Sensitivity and specificity values were determined by comparing the spectroscopic classification with histopathological examination [6, 35].
Receiver Operator Characteristic Analysis
Receiver operator characteristic (ROC) curve was plotted for fractional amplitudes (AI, AF and AS), component lifetime (τI, τF and τS ) and average lifetime (τavg) to determine the optimal cutoff value that would give the maximum sensitivity and specificity. The area under the curve (AUC) for each ratio variable was also calculated. The AUC must be close to 1 for maximum discrimination efficiency [6].
Results
Fluorescence Spectroscopic Characterization of Oral Tissues
Tryptophan has been used as the primary fluorescence probe in studying the protein in its native state. The averaged fluorescence emission spectra of oral tissues at 280 nm excitation were shown in Fig. 1. From the figure, it was observed that the normal tissue emission intensity was higher than that of malignant tissues and the broad emission band was observed in the wavelength region of 330 nm–350 nm. The minor humps were observed at 439 nm and 451 nm. It may be due to the presence of NADH and at 467 nm a hump may be due to lamp peak.

Averaged fluorescence emission spectra at 280 nm excitation of oral normal and cancerous tissue
In order to assess the spectral variations accurately, the normalized emission spectra of normal and malignant oral tissues were shown along with their difference spectrum in Fig. 2a. From the normalized emission spectra, it was observed that there was no shift in the spectrum with respect to normal and malignant oral tissues. The difference spectrum along with the normalized spectra shows a positive variation at 410 nm and negative variations at 335 nm and 460 nm respectively. In short, the spectral region at 330–350 nm which shows negative valley confirms the significant spectral variation in the fluorescence emission spectra.

(a) The normalized fluorescence emission spectra of normal and cancerous oral tissue with their difference spectrum and (b) the FWHM values of averaged fluorescence emission spectra for normal and cancer oral tissue at 280 nm excitation
The FWHM analysis was carried out for every individual recorded fluorescence emission spectra for 20 normal and 40 malignant tissues as shown in Fig. 2b. From the figure, it was observed that the FWHM value of normal tissues was higher than that of malignant tissues as their values were 86.87 ± 15.33 nm and 71.02 ± 16.36 nm respectively. The student’s t-test analysis reveals that the data were statistically significant (p = 0.001).
The averaged fluorescence excitation spectra of normal and malignant oral tissues were shown in Fig. 3a. The excitation spectra were recorded in the wavelength region of 250–330 nm by fixing the emission monochromator at 350 nm. From the spectra, it was observed that the normal tissues exhibit higher fluorescence emission intensity than that of malignant tissues and the peak at 293 nm may be attributed to absorption of tryptophan. The spectrum exhibits an isosbestic point at 302 nm and from 302 nm to 316 nm exhibits higher fluorescence emission intensity than that of normal. Subsequently, after 317 nm the normal dominates the malignant one. Further, a spectral shift was clearly observed in the excitation spectral profile. To account the spectral shift accurately, the excitation spectra were normalized and the corresponding difference spectra shown Fig. 3b.

(a) The averaged fluorescence excitation spectra and (b) normalized excitation with their difference spectra of normal and cancerous oral tissue at 350 nm emission
From the Fig. 3b, the normalized malignant excitation spectra shows a spectral shift of 7 nm with respect to normal tissues. This spectral shift can be attributed due to altered biochemical, morphological and conformational changes of the corresponding protein. The difference spectrum shows a positive peak in the region of 250–296 nm and a negative dip at 310 nm. Further, the difference spectrum at 296 nm and 321 nm show null difference. These spectral shifts clearly confirm the changes in the protein fluorescence due to the transformation of normal cells to malignant conditions.
To quantify and characterize the general features of the fluorescence emission spectral differences between normal and cancer subjects, two ratio variables I275/I310 and I310/I328 were introduced in the fluorescence excitation spectra at 350 nm emission. The mean and p value calculated using student’s t-test were shown in Table 1. The student’s t-test of both ratio variables exhibits higher significance with p value of 0.000. Further, the LDA was performed across 22 normal and 40 malignant subjects for the above two ratio variables using Leave-one-out Cross Validation method. From the Fig. 4a, it is observed that the ratio variable R1 discriminates the cancer oral tissue from their normal counterpart with 86.4 % specificity and 82.5 % sensitivity whereas the ratio variable R2 discriminates with 86.4 % specificity and 70 % sensitivity (Fig. 4b). Among the two ratio variables, it is further observed that R1 discriminates with better sensitivity and specificity than that of R2.

Scatter plot showing the distribution of Ratio Variables (R1 = I275/I310 and R2 = I310/I328)
Time Resolved Fluorescence Spectroscopy Characterization of Oral Tissues
The alteration in the protein conformation and local microenvironment could be analyzed with respect to its corresponding changes in the lifetime values and its amplitudes. In this regard, to probe the conformational changes in protein fluorescence, the fluorescence decay kinetics of oral tissues were obtained at 350 nm emission using 280 nm excitation (Fig. 5). The decay kinetics were fitted using the Decay Analysis Software (DAS). The mean, standard deviation and P value of fractional amplitudes, component lifetime and average lifetime are tabulated in Table 2.

Typical decay kinetics of normal and malignant oral tissues at 350 nm emission
Component Lifetime and Fractional Amplitudes
The intermediate component (τI) for normal tissue showed 2.22 ± 0.46 ns and malignant tissue showed 2.01 ± 0.58 ns. The average lifetime of normal tissue shows 0.21 ns difference with respect to malignant tissues and student’s t-test was not significant (p = 0.114). The fast lifetime component (τF) for normal tissues exhibit 0.88 ± 1.59 ns lifetime, which is shorter than that of the malignant tissues 1.30 ± 1.60 ns. The student’s t-test show that the data was not significant (p = 0.324). The slow component lifetime for normal tissue showed longer lifetime (τS = 6.50 ± 1.06 ns) than that of malignant tissues (τS = 6.06 ± 0.749 ns). The student’s t-test was computed with the p value of 0.061. Briefly, it was observed that normal tissue shows longer fluorescence decay time when compared to malignant tissues except for fast component lifetime (τF).
Fractional amplitudes of the corresponding lifetime for intermediate component amplitude (AI) of normal and malignant tissues were 33.02 ± 9.28 (%) and 31.03 ± 10.78 (%) respectively. It reveals that the equal contribution of photons was observed for normal and malignant tissues. The amplitude (AF) of the fast component was 33.77 ± 17.19 (%) for normal samples and 30.77 ± 19.5 (%) for its malignant counterpart. The amplitudes for both short and intermediate components were higher in normal than that of malignant tissues. For slow component (As) the amplitude for normal shows 33.21 ± 12.69 (%) and malignant show 38.20 ± 15.05 (%). The amplitude distribution of malignant show higher photon counts than that of normal. In contrast, third component amplitude shows reverse photonic distribution when compared to the intermediate and fast component amplitudes. The student’s t-test reveals that the fractional amplitudes were not statistically significant.
Changes in Average Lifetime
The average lifetime for normal samples exhibits longer lifetime (τN = 5.06 ± 0.31 ns) than that of malignant tissues (τM = 4.77 ± 0.23 ns) with the difference of ~0.30 ns and the student’s t-test show higher statistical significance (p = 0.001). Further, the component lifetime and amplitudes were analyzed using Receiver Operator Characteristics (ROC) curve. When compared with component lifetime and amplitudes, average lifetime reveals better sensitivity and specificity. Thus, the average lifetime may also be used to discriminate normal and malignant tissues.
Receiver Operator Characteristics (ROC) Curve Analysis
The component lifetimes (τI, τF and τS ) and fractional amplitudes (AI, AF and AS) were analysed using the ROC analytical tool (Table 3). Among photonic contributions of amplitude and lifetime, the average lifetime shows better sensitivity and specificity to discriminate cancer from normal. The average lifetime (τavg) shows 77.3 % Sensitivity and 70 % Specificity with the cut off value 4.85 ns (Fig. 6)

Receiver Operator Characteristics (ROC) curve for Component Lifetimes (τI, τF and τS), Fractional Amplitudes (AI, AF and AS) and average lifetime (τavg)of Oral tissues
Discussion
In cancer diagnosis, for the past few decades fluorescence spectroscopy has been considered as an effective tool to monitor the functional, morphological and structual characteristics of several endogenous fluorophores in tissues [4, 5]. Fluorescence spectroscopy was successfully applied in oral cancer diagnosis and achieved 80–100 % sensitivity with specificity [4]. During neoplastic progression in oral cavity show remarkable differences in spectral signature of the fluorophores. Among various endogenous fluorophores, due to its greater importance in natural proteins, tryptophan is one among the fluorophores which was extensively studied both theoretically and experimentally [9–12]. In this regard, the present study investigates the native fluorescence and time resolved fluorescence spectroscopy of normal and malignant oral tissues to probe the internal dynamics of protein under UV excitation.
Fluorescence emission spectra of oral tissues showed a major peak emission at 340 nm and it may be due to tryptophan. The reason for the fluorescence emission intensity of normal dominates the malignant tissues may be due to its biochemical and morphological changes in protein fluorescence on the prognosis of normal to malignant condition [4]. In addition to that, the FWHM of malignant tissues show narrow band emission spectra than that of normal counterpart. So, the FWHM value decreases when normal tissues prognosis to malignant condition. This may be attributed to the possible changes in concentration of amino acids present in tissues. This clearly indicates that the location of key amino acid, tryptophan changes with respect to its tissue transformation and particularly unfolding of proteins. The student’s t-test analysis of FWHM was statistically significant (P = 0.001).
The alteration in protein fluorescence can be effectily studied using fluorescence excitation spectroscopy. The averaged excitation spectra at 350 nm emission exhibits a major peak at 293 nm which may be due to the absorption of key amino acid tryptophan/tyrosine [5]. Previously, Yang et al. [36] reported that the excitation spectra of breast tissues show significant difference between normal and malignant subjects and also the peak in the excitation spectra was due to optical absorption of tryptophan/tyrosine. Similarly, Ganesan et al. [5] reported that the excitation spectra of normal and malignant epithelial cells also show that alteration in protein fluorescence.
Hence, the changes in the spectral intensity of normal and malignant oral tissues were due to changes in the optical absorption by amino acids and the proteins. Also variation in the concentration of tryptophan and its residues present in malignant tissues may be the reason for red shift in malignant tissues with respect to normal counter part [5]. The decreased absorption below 296 nm and increased absorption above 296 nm for malignant tissues when compared with normal cells indicates certain conformational and microenvironmental changes in the amino acid residues in proteins [5, 36]. Similar variation was observed in the difference spectrum which showed negative valley at 310 nm. The negative band (296–321 nm) indicates that the changes in the molecular environment in tissue protein.
Based on the above investigation, it was evident that the excitation spectroscopy of oral tissue serves as the indicator for the conformational changes in the cellular/tissue level of proteins. Further, LDA of two ratio parameters (R1 = I275/I310 and R2 = I310/I328) were introduced to discriminate malignant from normal tissues for statistical significance. Among them R1 provides better sensitivity and specificity of discriminating malignant from normal tissues. Thus the excitation spectra of oral tissues at 350 nm emission confirms the possible microenivronmental changes took place. This interesting fact leads to extend the study to obtain the decay kinetics of oral tissues in order to probe the conformational changes in the protein fluorescence under two different microenvironments and to study its excited state kinetics of key fluorophore.
Time Resolved Fluorescence Spectroscopy of Oral Tissues
Recent technological advancement in fluorescence lifetime reveals that time resolved fluorescence spectroscopy has been effectively considered by the medical community in order to obtain additional information owing to protein fluorescence. Although it has an added advantage in analyzing biochemical alterations of biological tissues and fluids accurately, to the best of our knowledge limited number studies were reported in oral tissues. Previously, Chen et al. [28] had investigated the normal and premalignant oral tissues in vivo using hand held optical probe at 633 nm emission. It was observed that the decay kinetics was fitted with two exponentials, hence two lifetimes were observed. The longer lifetime may be due to PPIX and shorter component lifetime not known accurately.
Meier et al. [29] studied the time-resolved fluorescence spectroscopy of Head and Neck Squamous Cell Carcinoma (HNSCC) at 337 nm excitation and reported that the lifetime values were differing between normal and cancerous tissue at 390 nm and 460 nm emission. Subsequently, the same group had also investigated the time resolved fluorescence spectroscopy of Hamster Buccal pouch animal tumour model [37] and reported that the lifetime at 460 nm was 1.44 ns for healthy mucosa, 1.25 ns for dysplasia, 1.28 ns for CIS and 1.34 ns for carcinoma. However, the lifetime values for 380 nm and 635 nm emission do not show any statistical significance.
To the best of our knowledge, it is the preliminary report on time resolved kinetics of in vitro oral tissues at 350 nm under UV excitation. In this context, the present study investigates normal and malignant oral tissues excited at 280 nm excitation (Nano LED 280 nm) and the emission was recorded at 350 nm. As reported previously, tissues under UV excitation was dominated by tryptophan than tyrosine. Tryptophan is extensively considered as one of the popular fluorescent probe to understand different conformational states and structural heterogeneity under different physicochemical conditions. This is because of its indole chromopore which is highly sensitive to the local environment makes much easier to study the local structure and its dynamics [9, 10]. Thus it is necessary to study the photophysical properties of tryptophan in oral cancer to obtain the internal dynamics of tryptophan. For a number of years, there has been a problem associating with the tryptophan lifetime in water as well as in proteins whether it is double or triple exponential.
In this regard, it is also reported that single tryptophan residues in a protein or peptide exhibits multi exponential decay and their corresponding amplitudes depend on emission wavelength. For example, the two fluorescent lifetimes of tryptophan in water at pH 7 is due to its dependence upon the structural complexity of tryptophan where the local environment of the indole chromophore also varies substantially. Hence, the heterogeneity was mainly associated with the three rotamer of tryptophan structure involving Cα and Cβ bonds related to long lived component of the indole excited state [9, 10]. Also, the shorter lifetime component may be due to rotamers in which the indole is closer to carboxyl group. The time resolved fluorescence emission of tryptophan in water undergoes multi-exponential decay even at room temperature and its lifetime value varies from 0.014 to 9.8 ns [9, 10].
In this way, the physical interpretation of multimodal distribution of tryptophan in its native state could be the indicator for various situation such as alteration in the rotamer state of tryptophan residue, changes in protein conformation of tryptophan residue due to variation in microenvironment or longer decay in microstate and fluctuation in the local environment of tryptophan residue in case of multi-tryptophan protein [10]. As tryptophan itself exhibits multi exponential decay in water, the analysis of decay kinetics of tryptophan in tissues makes it difficult to analyse the multiple lifetime correlation with tryptophan residues and its various conformational state [9–12].
In these circumstances, it is very difficult to monitor the changes in protein conformation exactly in its native state. Hence, the observed decay kinetics were fitted with three exponentials exhibiting three lifetimes and three fractional amplitudes. The decay kinetics justifies the tri-exponential fit as single or double show higher chi-square value. The observed three lifetimes may be due to different forms of tryptophan and its residues in different conformational states in tissues. Further, possible changes in rotamer state of tryptophan and it residues could be the reason for observed triple exponential. Thus, the intermediate and fast component lifetime may be attributed to the three conformers of zwitterion in tryptophan and the slow component lifetime may be associated with the tryptophan anion [8]. The fluctuation in fast component lifetime may be due to local pH and/or quenching mechanism observed in lifetime of the fluorophores [38]. Our results are well correlated with Asima Pradhan et al. [8] studies which investigates the metastatic and non metastatic cell lines under UV excitation.
Subsequently, the variation in the component lifetimes and their corresponding fractional amplitudes of normal were observed with respect to malignant tissues. The component lifetime and fractional amplitudes strongly depended on their microenvironment such as viscosity and chromophores of charged group [27]. As a single tryptophan and its residues conformers itself were strongly influenced by their microenvironment, thereby conformational changes possibly results in changes over lifetime. When the alteration of protein in malignant tissues changes subsequently the biochemical and morphological alteration leads to variation in normal and malignant tissue fluorescence lifetimes [6, 21]. As reported by Tata et al. [26], there could be the presence of two tryptophans in different proportions which may lead to alteration in average lifetime between normal and malignant tissues.
Further, the component lifetime and fractional amplitudes does not provide the statistical significance but the average lifetime exhibits statistical significance which is confirmed by the student’s t-test (P = 0.001). The ROC analysis of average lifetime results in 77 % sensitivity and 70 % specificity with the cut off value 4.85 ns. The component lifetime and fractional amplitudes were not statistically significant. Hence, the fluorescence lifetime spectroscopy technique may also be used in cancer diagnosis effectively. Further study is necessary to probe the exact reason for the variations in tryptophan lifetimes present in normal and malignant condition.
Conclusion
The photophysical properties of normal and malignant oral tissues under UV excitation were studied using native fluorescence and time resolved fluorescence spectroscopy. The difference in the FWHM values of fluorescence emission spectrum at 280 nm excitation for oral normal and cancer tissues exhibit statistical significant difference (P = 0.001). Further, the analysis of fluorescence excitation spectra confirms the alteration in the biochemical and morphological changes present in tissues. The two different ratio variables (R1 = I275/I310 and R2 = I310/I328) were introduced. Among them LDA of ratio variables reveals that R1 provides 86.4 % Specificity and 82.5 % Sensitivity. The analysis was extended to time resolved fluorescence spectroscopy of normal and malignant oral tissues exhibits that the average lifetime provides better significance when compared to fractional amplitudes and component lifetimes. The ROC analysis of average lifetime results in 77 % sensitivity and 70 % specificity with the cut off value 4.85 ns. In conclusion, over the component lifetime, average lifetime reads statistically significant. Further, the study can be extended to lifetime modeling, in order to probe the internal dynamics of protein.
References
Peterson PE (2008) Oral cancer prevention and control—The approach of the world Health Organization. Oral Oncol. doi:10.1016/J oraloncology.2008.05.023
Dikshit R, Gupta PC, Ramasundarahettige C et al (2012) Cancer mortality in India: A nationally representative survey. Lancet 379:1807–1816
Muller MG, Valdez TA, Georgakoudi I, Backman V, Fuentes C, Kabani S, Laver N, Wang Z, Boone CW, Dasari RR, Shapshay SM, Feld MS (2003) Spectroscopic detection and evaluation of morphologic and biochemical changes in early human oral carcinoma. Cancer 97:1681–1692
Ramanujam N (2000) Flurorescence spectroscopy of neoplastic and non-neoplastic tissues. Neoplasia 2:89–117
Ganesan S, Sacks PG, Yang Y, Katz A, Al-Ravi M, Savage HE, Schantz SP, Alfano RR (1998) Native fluorescence spectroscopy of normal and malignant epithelial cells. Cancer Biochem Biophys 16:365–373
Rajasekaran R, Aruna P, Koteeswaran D, Padmanabhan L, Muthuvelu K, Rai RR, Thamilkumar P, Murali Krishna C, Ganesan S (2013) Characterization and diagnosis of cancer by native fluorescence spectroscopy of human urine. Photochem Photobiol 89:483–491
Sivabalan S, Vedeswari CP, Jayachandran S, Koteeswaran D, Pravda C, Aruna P, Ganesan S (2010) In vivo native fluorescence spectroscopy and nicotinamide adinine dinucleotide/flavin adenine dinucleotide reduction and oxidation states of oral submucous fibrosis for chemopreventive drug monitoring. J Biomed Opt 15:017010-1-11
Pradhan A, Pal P, Durocher G, Villeneuve L, Balassy A, Babai F, Gaboury L, Blanchard L (1995) Steady state and time-resolved fluorescence properties of metastatic and non-metastatic malignant cells from different species. J Photochem Photobiol B 31:101–112
Alcala JR, Gratton E, Prendergast FG (1987) Fluorescence lifetime distributions in proteins. Biophys J 51:597–604
Swaminathan R, Krishnamoorthy G, Periasamy N (1994) Similarity of fluorescence lifetime distrivutions for single tryptophan proteins in the random coil state. Biophys J 67:2013–2023
Pan CP, Muiño PL, Barkley MD, Callis PR (2011) Correlation of tryptophan fluorescence shifts and lifetimes arising directly from heterogeneious environment. J Phys Chem B 115:3245–3253
Albani JR (2013) Origin of tryptophan fluorescence lifetimes part 1. Fluorescence lifetimes origin of tryptophan free in solution. J Fluoresc. doi:10.1007/s10895-013-1277-8
Ganesan S, Vengadesan N, Anbupalam T, Hemamalini S, Aruna P, Karkuzhali P (2002) Native fluorescence spectroscopy of cervical tissues: Classification by different statistical methods. Proc SPIE 4613:18–23
Glassman WS, Liu CH, Tang GC, Lubicz S, Alfano RR (1992) Ultraviolet excited fluorescence spectra from nonmalignant and malignant tissues of the gynecological tract. Lasers in Life Sciences 5:49–58
Ramanujam N, Mitchell MF, Mahadevan A, Thomsen S, Silva E, Richards-Kortum R (1994) Fluorescence spectroscopy: A diagnostic tool for cervical intraepithelial neoplasia (CIN). Gynecol Oncol 52:1–38
Alfano RR, Tang GC, Pradhan A, Lam W, Choy DSJ, Opher E (1987) Fluorescence spectra from cancerous and normal human breast and lung tissues. IEEE J Quantum Electron QE-23:1806–1811
Tang GC, Pradhan A, Alfano RR (1989) Spectroscopic differences between human cancer and normal lung and breast tissues. Lasers Surg Med 9:290–295
Ramanujam N, Mitchell MF, Mahadevan A, Thomsen S, Malpica A, Wright T, Atkinson N, Richards-Kortum R (1996) Spectroscopic diagnosis of cervical intraepithelial neoplasia (CIN) in vivo using laser-induced fluorescence spectra at multiple excitation wavelengths. Lasers Surg Med 19:63–74
Silveira L Jr, Betiol Filho JA, Silveira FL, Zângaro RA, Pacheco MT (2008) Laser-induced fluorescence at 488 nm Excitation for detecting benign and malignant lesions in stomach mucosa. J Fluoresc 18:35–40
Majumder SK, Gupta PK, Uppal A (1999) Autofluorescence spectroscopy of tissues from human oral cavity for discriminating malignant from normal. Lasers in Life Sciences 8:211–227
Svistun E, Alizadeh-Naderi R, El-Naggar A, Jacob R, Gillenwater A, Richards-Kortum R (2004) Vision enhancement system for detection of oral cavity neoplasia based on autofluorescence. Head Neck 26:205–215
Lakowicz JR (2006) Principles of fluorescence spectroscopy, 3rd edn. Springer, Singapore
Katz A, Ganesan S, Yang Y, Tang GC, Budansky Y, Celmer E, Savage HE, Schantz SP, Alfano RR (1996) Optical biopsy fiber based fluorescence spectroscopy instrumentation. SPIE, Proc. doi:10.1117/12.237579, 2679
Blackwell J, Katika KM, Pilon L, Dipple KM, Levin SR, Nouvong A (2008) In vivo time-resolved autofluorescence measurements to test for glycation of human skin. J Biomed Opt 13(1):014004
Albani JR (2013) Origin of tryptophan fluorescence lifetimes. Part 2: Fluorescence lifetimes origin of tryptophan in proteins. J Fluoresc. doi:10.1007/s10895-013-1274-y
Tata DB, Foresti M, Cordero J, Tomashefsky P, Alfano MA, Alfano RR (1986) Fluorescence polarization spectroscopy and time-resolved fluorescence kinetics of native cancerous and normal rat kidney tissues. Biophys J 50:463–469
Pradhan A, Das BB, Yoo KM, Cleary J, Prudente R, Celmer E, Alfano RR (1992) Time-resolved UV photoexcited fluorescence kinetics from malignant and non-malignant breast tissues. Lasers in Life Sci 4:225–234
Chen HM, Chiang CP, You C, Hsiao TC, Wang CY (2005) Time-resolved autofluorescence spectroscopy for classifying normal and premalignant oral tissues. Lasers Surg Med 37:37–45
Meier JD, Xie H, Sun Y, Sun Y, Hatami N, Poirier B, Marcu L, Farwell DG (2010) Time-resolved laser-induced fluorescence spectroscopy as a diagnostic instrument in head and neck carcinoma. Otolaryngol Head Neck Surg 142:838–844
Li Y, Li M, Xu T (2007) Quantitative time-resolved fluorescence spectrum of the cortical sarcoma and the adjacent normal tissue. J Fluoresc 17:643–648
Venius J, Bagdonas S, Žurauskas E, Rotomskis R (1996) Time-resolved fluorescence spectroscopy of the heart tissues. Lith J Phys 51:370–376
Alfano RR, Tang GC, Pradhan A, Bleich M, Choy DSJ, Opher E (1988) Steady state and time-resolved laser fluorescence from normal and tumor lung and breast tissues. J of Tumor Marker Oncology 3:165
Ebenezar J, Aruna P, Ganesan S (2010) Synchronous fluorescence spectroscopy for the detection and characterization of cervical cancers in vitro. Photochem Photobiol 86:77–86
Rajasekaran R, Aruna P, Balu David M, Koteeswaran D, Muthuvelu K, Rai R, Ganesan S (2013) Steady state and time-resolved fluorescence spectroscopic characterization of normal and cancerous urine. Proc SPIE. doi:10.1117/12.2006086
Madhuri S, Vengadesan N, Aruna P, Koteeswaran D, Venkatesan P, Ganesan S (2003) Native fluorescence spectroscopy of blood plasma in the characterization of oral malignancy. Photochem Photobiol 78:197–204
Yang Y, Katz A, Celmer EJ, Zurawska-Szczepaniak M, Alfano RR (1997) Fundamental differences of excitation spectrum between malignant and benign breast tissues. Photochem Photobiol 66:518–522
Farwell DG, Meier JD, Park J, Sun Y, Coffman H, Poirier B, Phipps J, Tinling S, Enepekides DJ, Marcu L (2010) Time-resolved fluorescence spectroscopy as a diagnostic technique of oral carcinoma: Validation in the hamster buccal pouch model. Arch Otolaryngol Head Neck Surg 136:126–133
Bouchard A, Fréchette J, Vernon M, Cormier JF, Beaulieu R, Vallée R, Mafu AA (2006) Optical characterization of pseudomonas fluorescens on meat surfaces using time resolved fluorescence. J Biomed Opt 11:014011
Acknowledgments
This work was supported by DAE-BRNS Grant (Ref. No. 2009/34/38), Govt. of India. The authors, K.U and M.Y acknowledge DST-PURSE for fellowship. We acknowledge Dr. S. Sri Priya, Mrs. Arivazhagi of Government Arignar Anna Cancer Hospital, Regional Cancer Centre, Karapettai, Kanchipuram, for their constant support and providing samples for our experiments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Udayakumar, K., Yuvaraj, M., Awad, F. et al. Native Fluorescence and Time Resolved Fluorescence Spectroscopic Characterization of Normal and Malignant Oral Tissues Under UV Excitation—an In Vitro Study. J Fluoresc 24, 613–623 (2014). https://doi.org/10.1007/s10895-013-1335-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-013-1335-2