
Abstract
Puerarin is a widely used compound in Chinese traditional medicine and exhibits many pharmacological activities. Binding of puerarin to human serum albumin (HSA) was investigated by ultraviolet absorbance, fluorescence, circular dichroism and molecular docking. Puerarin caused a static quenching of intrinsic fluorescence of HSA, the quenching data was analyzed by Stern–Volmer equation. There was one primary puerarin binding site on HSA with a binding constant of 4.12 × 104 M−1 at 298 K. Thermodynamic analysis by Van Hoff equation found enthalpy change (\(\Delta H^{O} \)) and entropy change (\(\Delta S^{O} \)) were −28.01 kJ/mol and −5.63 J/mol K respectively, which indicated the hydrogen bond and Van der waas interaction were the predominant forces in the binding process. Competitive experiments showed a displacement of warfarin by puerarin, which revealed that the binding site was located at the drug site I. Puerarin was about 2.22 nm far from the tryptophan according to the observed fluorescence resonance energy transfer between HSA and puerarin. Molecular docking suggested the hydrophobic residues such as tyrosine (Tyr) 150, Tyr 148, Tyr 149 and polar residues such as lysine (Lys) 199, Lys 195, arginine 257 and histidine 242 played an important role in the binding reaction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
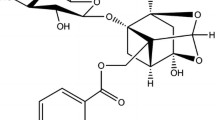
Flavonoids and isoflavonoids, two large groups of naturally occurring polyphenolic compounds, exhibit many bioactivities. Puerarin, an isoflavonoid C-glycoside isolated from pueraria lobota, is traditionally used as an anti-inebriation drug to reduce the febrile symptoms in China (Fig. 1). It has been shown to possess many pharmacological activities such as the antioxidant activity [1, 2], the protective activity against cardiovascular diseases and ischemic retinopathy [3, 4], and the antihyperglycemic activity [5]. More recent investigations reported puerarin could induce the apoptosis of cancer cell and attenuate the increase of total cholesterol [6, 7].

Structure of puerarin
Human serum albumin (HSA), the most abundant protein in plasma, acts as a transporter and disposer of many endogenous and exogenous compounds [8–13]. HSA is a single chain of 585 amino acids and folded into three homologous domains each of which contains two subdomains (A and B). It has two primary binding sites, site I and site II, which are hydrophobic cavities located in subdomain IIA and subdomain IIIA respectively. Most compounds bind to these two sites with an affinity constant of 104 ~ 106 M−1 [14, 15]. It is widely accepted that the distribution, metabolism, and efficacy of many drugs can be altered based on their binding to serum albumin [16].
Although the pharmacokinetics of puerarin in human plasma has been measured [17], there still has been little information about the interaction between puerarin and protein so far. To better understand the pharmacological activities of puerarin at molecular level, we characterized the interaction between puerarin and HSA by different spectroscopic methods as our previous studies [18, 19]. Synchronous fluorescence and circular dichroism (CD) measurement were employed to probe conformational changes induced by puerarin. Binding parameters were calculated according to fluorescence data, binding mode was discussed based on thermodynamic analysis. The precise location of puerarin on HSA was identified by competitive binding experiments, and was further calculated by fluorescence resonance energy transfer (FRET) and molecular docking.
Materials and methods
Materials and solutions
Human serum albumin (fatty acid free, 96 ~ 99%) were purchased from Sigma Chemical Co. (St. Louis, MO, USA); bovine serum albumin (BSA), Tris-base were from Amresco; puerarin (>99.0%) was from Nan Yang Zhangzhongjing Digital Chinese Medicine Co., Ltd. (Xi’xia, China); warfarin (>99.0%) was obtained from Wuhan Shiji Pharmaceutical Co. Ltd. (Wuhan, China); NaH2PO4, Na2HPO4 (analytic purity) were purchased from Shanghai Chemical Co.(Shanghai, China); ultrapure water from Mini Q was used for solution preparation. Other chemicals were of analytic grade without further purification.
Phosphate buffer solution (PBS) and Tris-Hcl buffer (50 mM) were adjusted with NaOH and HCl solution to different pH values at 298 K. NaCl of 0.1 M was added into the buffers to keep the ionic strength. HSA was firstly dissolved in PBS as stock solution, and then mixed with drug in PBS by micro-injector to obtain different HSA-drug ratios. Puerarin were dissolved in ethanol and then diluted with the phosphate buffer to obtain required concentrations. The final concentrations of ethanol were below 1% (V/V) for all spectroscopic measurements.
Apparatus and methods
Fluorescence spectra of HSA were recorded from 300–400 nm on F-4500 fluorescence spectrometer (Hitachi, Japan) at an excitation wavelength of 295 nm with different concentration of drug as reference. Excitation and emission bandwidth were 5 nm. Concentration of HSA was maintained at 3 × 10–6 M in PBS (pH 7.4) and ranged from zero to ten. To investigate the influence of pH on the binding of drug to HSA, the spectra of HSA were collected in PBS of pH 6.4, 7.4, 8.0 individually; and spectra of HSA in PBS (pH 7.4) were collected at the temperature of 298 K, 303, 310 and 315 K for examining the effect of temperature. Synchronous fluorescence spectra of HSA were also collected with the same concentration to probe the conformational changes of the protein. In fluorescence measurements, 2 ml PBS containing 3 × 10−6 M HSA were firstly added into 1cm cell, then successive puerarin (3 × 10−5 M) were injected into HSA with 2 μl very time to obtain different ratios of C puerarin/C HSA.
The ultraviolet (UV) absorption spectra of puerarin at the concentration of 3 × 10−5 M in Tris-Hcl buffer of different pH were recorded on a Cary 100 UV−visible spectrometer (Varian, USA) from 200–450 nm. Absorption spectra of HSA after mixed with puerarin were collected form 350–190 nm with the same concentration of HSA as previously.
Circular dichroism spectra were measured on a JASCO J-820 automatic spectropolarimeter (Jasco, Tokyo, Japan) using a 300 μl cell. Data were recorded from 200–250 nm with a scan speed of 100 nm/min. HSA were incubated with PBS containing different concentration of puerarin before the measurement, and the concentration of HSA was maintained at 3 × 10−6 M. All the spectral measurements were performed in PBS with pH 7.4 at 298 K unless explained in article, every measurement was repeated at least three times.
The program AutoDock 3.05 was used in this docking study, in which Lamarckian genetic algorithm was used to search for the optimum binding site [20]. For more accurate docking, we modified the parameters, which were 150 for ga_pop_size, 10,000,000 for ga_num_evals. During docking process, a maximum of 50 conformers was considered for the drug. The conformer with the lowest binding free energy was used for further analysis. The crystal structure of HSA entitled 1H9Z was taken from the Brookhaven Protein Data Bank (http://www.rcsb. org/pdb) [21], the 3D structure of puerarin was constructed by ChemOffice 2004, the docking result was rendered by POV-Ray version 3.20.
Results and discussion
Change of HSA secondary structure induced by puerarin
Figure 2 shows the UV absorption of HSA in the presence and absence of puerarin. The influence of puerarin was eliminated by using corresponding concentration of puerarin as reference solution during the measurement. As can be seen from the figure, HSA has two absorption peaks at 209 and 280 nm. The absorption of HSA at 210 nm represents the content of α-helix in the protein [22]. Addition of puerarin to HSA resulted in a decrease of absorbance intensity at 209 nm and a subtle change at 280 nm, which indicated a perturbation of α-helix induced by a specific interaction between puerarin and HSA.

UV absorbance of HSA with different concentrations of puerarin, C HSA = 3.0 × 10−6 M, from (a) to (d), the value of C puerarin/C HSA were 0, 1, 10, 20 respectively, (e) shows the blank control, pH = 7.40, T = 298K
To ascertain the possible influence of drug binding on the secondary structure of HSA, we have performed CD studies in the presence of different concentrations of puerarin. As shown in Fig. 3, the CD spectra of HSA had two negative binds in the far UV region at 208 and 220 nm, which are typical characterization of α-helix structure in protein [23]. The reasonable explanation is that the negative peaks between 208 and 209 and 222–223 nm are contributed to n−π* transfer for the peptide bond of α-helical [24]. Addition of puerarin to HSA caused only a subtle decrease in negative ellipticity in the region of far-UV CD without any significant shift of peaks, indicating that the binding of drug to HSA induced a slight decrease in the α-helical structure content of the protein. The similarity between the CD spectra shapes of free HSA and puerarin-HSA suggested structure of HSA was also predominantly α-helix. The α-helix contents of free and combined HSA were calculated from mean residue ellipticity values at 222 nm using the flowing equation [25]:

CD spectra of puerarin–HSA in the absence and presence of increasing amount of puerarin, the ratio of C puerarin/C HSA = 0,1,5 (a, b, c); C HSA = 3.0 × 10−6 M; pH = 7.4; T = 298 K
Where n is the number of residues in protein, l is the path length, C P is the concentration of protein. According to the calculated results, the α-helix contents of HSA were 47.03% and 46.56% with the molar ratio puerarin–HSA of 0:1 and 1:1 respectively, it retained at 45.91% even the ratio was up to 5:1. Then it suggested that binding of puerarin to HSA had not alter the secondary structure of the protein significantly. Previous study on anaesthetic binding to HSA showed that occupancy of the tryptophan sites by the interacting ligands actually stabilized the native conformation of HSA, and it was suggested that the polarity of the ligand was an important driving force for the interaction [26]. Some other flavonoids were also found to bind tightly to protein without changing its secondary structure, which agrees the result reported here [27]
Conformational changes investigated by synchronous fluorescence
Synchronous fluorescence is a very useful tool to investigate the microenvironments around the fluorophore functional groups. It is well known that the fluorescence of HSA arises from the tyrosine, tryptophan and phenylalanine residues. According to Miller [28], the fluorescence of HSA with \(\Delta \lambda {\left( {\Delta \lambda = \lambda _{{{\text{emission}}}} - \lambda _{{{\text{exitation}}}} } \right)}\) of 60 and 15 nm are characteristic of tryptophan and tyrosine respectively. The effect of puerarin on synchronous fluorescence spectra was shown in Fig. 4. It can be seen the fluorescence spectra of tyrosine (Fig. 4b) was weak and addition of puerarin resulted in a decrease in intensity and no shift of maximum emission wavelength (λ max). The fluorescence of tryptophan (Fig. 4a) was strong, with addition of puerarin the fluorescence intensity decreased and the λ max shifted form about 338 to 344 nm. It is reported that maximum emission wavelength at 330–332 nm indicates the tryptophan residues are located in an apolar region, that means they are buried in a hydrophobic cavity; and the λ max at 350–352 nm shows tryptophan residues are exposed to water, namely, the hydrophobic cavity in HSA is disagglomerated and the structure of HSA is looser [28]. So the observed red shift of λ max suggested puerarin bound to a hydrophobic cavity in HSA, the polarity around tryptophan increased while the hydrophobicity decreased.

Synchronous fluorescence spectra of HSA with Δλ = 60 nm (a) and Δλ = 15 nm (b) in the absence and presence of increasing amount of puerarin. The ratio of C puerarin/C HSA were 0,1,2, 4,5,6, 8 and 10; C HSA = 3.0 × 10−6 M; pH = 7.4; T = 298 K
Analysis of fluorescence quenching of HSA by puerarin
The sole tryptophan (Trp)-214 residue located in the subdomain II of HSA makes fluorescence spectroscopic measurement the most popular tool to examine the binding of different ligands to HSA, because only the tryptophan residue has florescence emission when the excitation is 295 nm. The fluorescence quenching spectra of HSA in presence of different concentrations of puerarin were shown in Fig. 5(a). Addition of puerarin caused a dramatic change in the emission spectra. The fluorescence intensity decreased gradually with the increase in concentration of puerarin, and higher concentrations led to more efficient quenching of the protein fluorescence. Such a strong quenching clearly indicated the binding of puerarin to HSA. The change of microenvironment around tryptophan was also testified by a blue shift of maximum emission wavelength from 336 to 330 nm.

(a) The fluorescence emission spectra of puerarin–HSA in the absence and presence of increasing amount of puerarin, the ratio of C puerarin/C HSA = 0, 0.5,1,2,3,4,5,6,7,8,9,10 from the (a) to the (l); C HSA = 3.0 × 10−6 M; λex = 295 nm, λem = 336 nm, pH = 7.4, T = 298 K. (b) Stern–Volmer curve of protein fluorescence quenching
Stern−Volmer equation was applied to analyze the mechanism of fluorescence quenching induced by puerarin [11, 29].
F 0 and F are the fluorescence intensities in the absence and presence of puerarin. Q is the drug concentration, and K SV is the Stern–Volmer quenching constant which is equal to K Q∙τ 0, K Q and τ 0 represent bimolecular quenching rate and the lifetime of fluorophore in the absence of drug.
As shown in Fig. 5 (b), the curve of F 0/F versus [Q] was plotted according to Eq. (1) and was with a good linearity when the ratio of C puerarin/C HSA ranged from 0.5 to 10. Then the regression equation was obtained, y=(0.99535 ± 0.02007) + (0.03888 ± 0.00113) × 106 Q. According to Eq. (1), the slope of the regression curve is the Stern–Volmer quenching constant. K SV=(3.88 ± 0.11) × 104 M−1∙s. The average fluorescence lifetime of HSA was about 5ns according to previous reports [29]. Then K Q, the bimolecular quenching rate, was calculated by the Eq. (1), K Q = 7.78 ± 0.23 × 1012 M−1∙s. The value of K Q was about 400 folds higher than limiting diffusion constant K diff of the biomolecules (K diff = 2.0 × 1010 M−1∙s), suggesting a static quenching mechanism by the specific interaction of puerarin with HSA.
To get the binding constant, quenching data from the fluorescence titration were analyzed according to the modified Stern–Volmer equation [30]:
F 0, F and Q are the same as Eq. (3), f is the fractional maximum fluorescence intensity of the protein summed up, K represents Stern–Volmer quenching constant, which is very close to binding constant of the molecule. The result showed the K for puerarin was (4.12 ± 0.03) × 104 M−1 at 298 K.
Thermodynamic analysis and the binding force
The ligand-binding process during protein–ligand complex is mainly governed by four types of weak, noncovalent forces including hydrogen bond, van der Waals force, electrostatic, and hydrophobic interactions. Elucidating the role of these interactions provides insights into the mechanism of molecular recognition. To characterize the acting forces between puerarin and HSA, thermodynamic parameters depended on temperatures were analyzed. Figure 6 (a) is the Stern–Volmer quenching plots of puerarin with HSA at different temperatures (298, 303, 310, 315 K). There was a linear dependence between F 0/(F 0−F) and 1/Q and the slopes increased with increasing temperature, which was indicative of the homogeneity of static quenching [31].

(a) The modified Stern–Volmer plots of the puerarin–HSA at different temperatures (filled square, 298 K; filled circle, 303 K; filled upright triangle, 310 K; filled inverted triangle, 315 K); (b) Van’t Hoff plot for the interaction of HSA and puerarin in PBS
If the enthalpy change (\(\Delta H^{0} \)) does not vary significantly in the temperature range studied, its value and that of entropy change (\(\Delta S^{0} \)) can be calculated from the Van’t Hoff equation [30]:
K is the binding constant at corresponding temperature T, R is the gas constant, \(\Delta G^{0} \) represents free energy.
According to the binding affinity constants of puerarin to HSA obtained at the four temperatures above, the thermodynamic parameters determined from linear Van’t Hoff plot (Fig. 6 (b)) were presented in Table 1. The negative \(\Delta G^{0} \) (−28.01 kJ/mol) suggested the binding of puerarin with HSA was a spontaneous process. \(\Delta H^{0} \) and \(\Delta S^{0} \) for the binding reaction were −28.01 kJ/mol and −5.63 J/mol K, which revealed that the binding process was an exothermic reaction accompanied by negative \(\Delta S^{0} \) value. Ross and Subramanian [32] have characterized the sign and magnitude of the thermodynamic parameter associated with various individual kinds of interaction which may take place in protein association processes. From the point of view of water structure, a negative \(\Delta S^{0} \) value is often contributed by van der Waals interaction or hydrogen bond. Furthermore, specific electrostatic interactions between ionic species in aqueous solution are characterized by a positive value of \(\Delta S^{0} \) and a negative \(\Delta H^{0} \). Accordingly, it was impractical to attribute the formation of puerarin–HSA complex to a single intermolecular force. Evidenced by a negative \(\Delta S^{0} \), it was more likely that the hydrogen bond and van der Waals interaction had played a significant role in the binding reaction. The large negative \(\Delta H^{0} \) probably mainly came from electrostatic interactions. The major contribution to \(\Delta G^{0} \) arose from the \(\Delta H^{0} \) term rather than from \(\Delta S^{0} \) meant the binding process was driven by enthalpy and the hydrogen bond was most likely to be the predominant force in the binding process.
Identification of binding site
The number of binding site was calculated according to flowing equation deduced in previous investigations [11].
There was a good linearity of regression curve based on Eq. (4) in the studied range (0.5–10) of C puerarin/C HSA, and the slope of the regression curve (number of binding site) was 0.962, which meant there was only one puerarin binding site in the HSA molecule.
In order to identify the binding site for puerarin on HSA, fluorescence quenching with defatted BSA was measured. HSA and BSA have similar folding with a well-known primary structure, however, there is an important difference between them: BSA has two tryptophan residues (Trp-134 and Trp-212) located in domain I and domain II respectively, while HSA has only one tryptophan at position 214 in domain II. This property can be used to identify the binding pocket for drugs in HSA [12]. As shown in Fig. 7 (B) , the quenching curve of HSA was almost the same as that of BSA in spite of their differences, the overlap of the mass action plots showed the similar binding site of puerarin to the two proteins. BSA tryptophan residue located at position 212 is in analogy to the single tryptophan of HSA located at 214, the quenching of protein fluorescence was probably due to accommodation of puerarin near these two tryptophans. In another words, the binding site of puerarin was probably located in domain II B (site I). In the case of BSA, the fluorescence from Trp 212 can be quenched by puerarin in the same way as that of Trp 214 in HSA; Trp 134 is very close to Trp 212, binding of puerarin to site I may therefore quench the fluorescence form both tryptophans of BSA. Moreover, binding of puerarin changed the geometry of HSA and facilitated quencher penetrating into protein interior, the penetration may also result in quenching of fluorescence from Trp 134 in BSA.

(A) Titration of warfarin–HSA by puerarin with concentrations from 0 to 3.0 × 10−5 M μm, (a) to (k), (i) represents the spectra of HSA, C HSA = C warfarin = 3.0 × 10−6 M, λex = 320 nm; (B) Fluorescence quenching of HSA (filled circle), free BSA (filled triangle), and warfarin–HSA (circle) by puerarin, pH = 7.4, T = 298 K
Crystallographic analysis has shown that warfarin bond to the drug site I of HSA [13], so we used warfarin as a site marker to perform the competition experiment. When excited at 320 nm, warfarin–HSA shows a strong emission from 330 ~ 500 nm, while free warfarin does not [33], then the titration of HSA–warfarin complex was performed to investigate the displacement. As demonstrated in Fig. 7 (b), adding puerarin to HSA–warfarin mixture resulted in a decrease in the fluorescence intensity and a red shift of maximum emission wavelength from 351 to 356 nm. Then the quenching of HSA–warfarin was analyzed by Stern–Volmer plot (Fig. 7 (b)), the good linearity of these data indicated a static quenching mechanism was dominant. This could be due to displacement of warfarin by puerarin. The moderate quenching rather than complete quench indicated partial overlap between these their binding sites. The red shift of λ max suggested an increase polarity around warfarin, a reasonable explanation was that some of the bound warfarin fell out of the binding site and exposed to water [34]. That was to say, binding of puerarin led a disruption of HSA–warfarin complex. Thus, it can be concluded that the binding site of puerarin was located in drug site I with partial rather than complete overlap with warfarin binding site.
Energy transform between puerarin and HSA
In order to determine the precise location of puerarin in HSA, fluorescence resonance energy transfer was used. FRET is a distance dependent interaction between the different electronic excited states of dye molecules in which excitation energy is transferred from one molecular (donor) to another molecular (acceptor) without emission of a photon from the former molecular system. According to Föster’s theory [35], the efficiency of FRET is mainly governed by three factors: (1) the extent of overlap between the donor emission and the acceptor absorption, (2) the orientation of the transition dipole of donor and acceptor, (3) the distance between the donor and the acceptor.
From Fig. 8, it was clearly seen that puerarin bound to HSA caused a decrease in tryptophan fluorescence of HSA, simultaneously this decrease was followed by a little increase in fluorescence due to puerarin at 575 nm. This suggested an energy transfer between excited tryptophan and the bound puerarin. Then the efficiency of FRET (E) can be given in terms of the distance between the donor and acceptor by following equation [36]:

(A) Fluorescence of free HSA (a), puerarin (c) and HSA-puerarin (b and b′). λex = 295 nm; (B) spectral overlap of HSA fluorescence (a) with puerarin absorbance (b), C puerarin = C HSA = 3.0 × 10−6 M, pH = 7.4, T = 298 K
F 0 and F are the fluorescence intensity of donor in the absence and presence of equal amount of acceptor respectively, r 0 is the distance between acceptor and donor and R 0 is the critical distance when the transfer efficiency reaches 50%, and the value of R 0 is calculated by following equation:
Where K 2 is the spatial orientation factor of the dipole, n is the refractive index of the medium, Φ is the fluorescence quantum yield of the donor and J is the overlap integral of the fluorescence emission spectrum of the donor and the absorption spectrum of the acceptor. J was given by:
In this equation F(λ) is the fluorescence intensity of the fluorescent donor of wavelength, λ, ε(λ) is the molar absorption coefficient of the acceptor at wavelength, λ. In the present case, K 2 = 2/3, n = 1.36 and Φ = 0.074 [9]. Figure 8 shows the overlap of UV absorbance spectrum of puerarin with fluorescence spectrum of HSA. According to Eq. (8)~Eq. (10), we calculated the value of different parameters: J =(8.86 ± 0.04)×10−16 cm3 l/mol, R 0 = 1.50 ± 0.01 nm, r 0 = 2.22 ± 0.05 nm. The average distance between the a donor fluorophore and acceptor fluorophore is on scale of 2–8 nm, and 0.5R 0 < r 0 < 1.5R 0 indicated a high possibility of energy transfer between HSA and puerarin, while r 0 was bigger than R 0 in this study also revealed that puerarin could strongly quench the intrinsic fluorescence of HSA by non-radiative energy transfer and static quenching [37].
Effect of pH on binding of puerarin to HSA
To better understand the binding mode between puerarin and HSA, we also examined the influence of pH on the binding reaction. Firstly we characterized the UV absorbance of puerarin in different pH as shown in Fig. 9 (a) . Like other flavonoids, the UV spectra of puerarin exhibit two major absorption bands, band-I (300–380 nm) and bind-II (200–280 nm), which are associated with absorption of ring-B cinnamoyl and ring-A benzoyl system [38]. The absorption peak at 306 nm gradually disappeared and a new peak at about 345 nm rose as pH increased, and the peak at 248 nm shifted to about 258 nm. These changes of the absorption spectra were considered to be related to disproportionation of two hydroxyl at position 4′ and position 7. The prokineticin 1 (PK1) and PK2 for these to hydroxyl were reported to be 9.93 and 6.91 respectively [39]. The puerarin is mainly ionized and with two negative charges on the molecule in the basic environment, partially ionized and with one negative charge at neutral condition, and with no ionization and charge in acidic solution.

(A) The UV absorbance spectra of puerarin in different pH buffers, the pH of buffer are (a)–(g) 5.74, 5.92, 6.41, 6.91, 7.40, 8.01, 8.48. C puerarin = 3 × 10−5 M; (B) the Stern–Volmer plots of the quenching of HSA fluorescence by puerarin at pH of 6.4 (filled triangle), 7.4 (filled square) and 8.0 (filled circle). C HSA = 3.0 × 10−6 M; T = 298 K
Figure 9 (b) displays the Stern–Volmer plots of the quenching of HSA tryptophan residues fluorescence by puerarin at pH of 6.4, 7.4 and 8.0, the calculated binding affinities are listed in Table 2. The binding affinity for puerarin to HSA increased with the increasing pH value of PBS, which indicated that the ionized puerarin was more favorable to HSA. HSA is known as undergo several different pH dependent transitions, the N–F transition between 7.0 and 3.5, the F–E transition between 3.5 and 1.2, and the N–B transition between 7.0 and 9.0 [40]. At pH 6.4, HSA essentially existed as N conformation, while the decreasing binding affinity showed that the binding process was probably dominated by the drug in its ionized state. At pH 8.0, HSA is changing into the B conformation. The B isomerization is supposed to be a structural fluctuation, a loosening of the molecule with loss of rigidity, probably affecting the ligand binding. Moreover, puerarin was almost completely ionized in higher pH with two negative charges, these charges could probably interact with basic residues in HSA then strengthen the binding [41].
Molecular docking of the puerarin–HSA complex
Taking of all previous data together, we inferred that puerarin bound to the drug site I located in subdomain II A. Molecular docking search in and around the warfarin binding site identified a site suitable for puerarin. Figure 10 showed the most possible binding site (30/50) of puerarin computed by AutoDock 3.05, which was suited approximately 8 Å far from warfarin. The distance between Trp and binding site was about 16.7 Å which was close to that from FRET calculation (2.22 nm), it confirmed that the binding site was located in drug site I. Puerarin molecule was suited in a cavity formed by lysine (Lys) 106, Tyr 150, Lys 195, His 242 and arginine (Arg) 257. The B ring was located in a very hydrophobic pocket formed by Tyr 150, Tyr 148, Tyr 149, which indicating a hydrophobic interaction between these phenyl rings. The coplanar A ring and C ring were fixed in a bigger but less hydrophobic part of the binding chamber, and the glycosyl was exposed to the entrance of the binding site. The 4′-OH and 7-OH were making a hydrogen bond with mainchain carbonyl oxygen of glycine 248 and Lys 195 respectively, another hydrogen bond was formed between the −OH of glycosyl and mainchain carbonyl oxygen of serine 192, water molecules were also likely to serve as a bridge to produce some other hydrogen bonds. It was also important noted that many basic residues such as Lys 106, Lys 199, Lys 195, Arg 257 and His 242 were very in close proximity to the puerarin, this suggested a strong electrostatic interaction also existed in the binding process. Combined with the thermodynamic analysis, we believed that hydrogen bonds and electrostatic interactions are the driven forces rather than the hydrophobic interaction, a reasonable explanation was that glycosyl caused an increase in hydrophilicity of puerarin, and weakened the hydrophobic interactions. Earlier reports have already found addition of a glycosyl to the aromatic rings resulted in a decrease of binding affinity [10]. Recent investigation about binding of daidzein also got higher binding affinity than puerarin, while the difference of these two compounds is only that puerarin has a glycosyl on position 8 [12].

Molecular docking of the puerarin–HSA complex
Conclusions
Binding interaction of puerarin with HSA has been investigated in vitro under a simulated physiological condition (ionic strength = 0.1 mol/l, pH 7.4) by different spectroscopic methods. The UV absorbance and CD spectra suggested binding of puerarin to HSA induced a slight decrease in α-helical contents. The microenvironment around HSA also became less hydrophobic according to data from synchronous fluorescence. Static quench of HSA intrinsic fluorescence by puerarin also suggested a complication of HSA with puerarin. Thermal dynamics analysis elucidated the binding reaction was mainly driven by hydrogen bond, van der Waals interaction and electrostatic interaction. The binding constant increased with the increasing pH value implied the hydroxyl of puerarin play a significant role in the binding process. Competitive binding experiments with warfarin indicated the binding site was located in a hydrophobic cavity near the drug binding site I.
References
Zhu QL, He AX, Lu XR (2001) Effects of puerarin on the scavenge of oxygen free radicals and the antagonism against oxidative injury. Pharm J Clin PLA 17:1–4
Xu XH (2003) Effects of puerarin on fatty superoxide in aged mice induced by D-galactose, Chin. J Chin Mater Med 28:66–69
Benlhabib E, Baker JI, Keyler DE, Singh AK (2004) Effects of purified puerarin on voluntary alcohol intake and alcohol withdrawal symptoms in P rats receiving free access to water and alcohol. J Med Food 7:180–186
Xuan B, Zhou YH, Yang RL (1999) Improvement of ocular blood flow and retinal functions with puerarin analogs. J Ocular Pharmacol Ther 15:207–216
Hsu FL, Liu IM, Kuo DH, Chen WC, Su HC, Cheng JT (2003) Antihyperglycemic effect of puerarin in streptozotocin-induced diabetic rats. J Nat Prod 66:788–792
Yu ZL, Li WJ (2006) Induction of apoptosis by puerarin in colon cancer HT-29 cells. Cancer Lett 238:53–60
Yan LP, Chan SW, Albert Sun-Chi Chan, Chen SL, Ma XJ, Xu HX (2006) Puerarin decreases serum total cholesterol and enhances thoracic aorta endothelial nitric oxide synthase expression in diet-induced hypercholesterolemic rats. Life Sci 79:324–330
Lu Z, Zhang Y, Liu H, Yuan J, Zheng Z, Zou G (2007) Transport of a cancer chemopreventive polyphenol, resveratrol: interaction with serum albumin and hemoglobin. J Fluoresc 17(5):580–587
Hu YJ, Liu Y, Zhang LX (2005) Study of interaction between colchicines and bovine serum albumin by fluorescence quenching method. J Mol Struct 750:174–178
Dufour C, Dangles O (2005) Flavonoid-serum albumin complexation: determination of binding constants and binding sites by fluorescence spectroscopy. Biochim Biophys Acta 1721:164–173
Xie MX, Long M, Liu Y, Qin C, Wang YD (2006) Characterization of the interaction between human serum albumin and morin. Biochim Biophys Acta 1760:1184–1191
Mahesha HG, Sighn SA, Srinivasan N, Appu RAG (2006) A spectroscopic study of the interaction of isoflavones with human serum albumin. FEBS J 273:451–467
Petitpas I, Bhattacharya AA, Twine S, East M, Curry S (2001) Crystal structure analysis of warfarin binding to human serum albumin—anatomy of drug site I. J Biol Chem 276:22804–22809
He XM, Carter DC (1992) Atomic structure and chemistry of human serum albumin. Nature 358:209–215
Dockal M, Chang M, Carter Rüker DCF (2000) Five recombinant fragments of human serum albumin—tools for the characterization of the warfarin binding site. Protein Sci 9:1455–1465
Dubois BW, Cherian SF, Evers AS (1993) Volatile anesthetics compete for common binding sites on bovine serum albumin: a 19F-NMR study. Proc Natl Acad Sci USA 90:6478–6482
Penetar DM, Teter CJ, Ma Z, Tracy M, Lee DY, Lukas SE (2006) Pharmacokinetic profile of the isoflavone puerarin after acute and repeated administration of a novel kudzu extract to human volunteers. J Altern Complement Med 12:543–548
Tang K, Qin YM, Lin AH, Hu X, Zou GL (2005) Interaction of daunomycin antibiotic with human serum albumin: investigation by resonant mirror biosensor technique, fluorescence spectroscopy and molecular modeling methods. J Pharm Biomed Anal 39:404–410
Yuan JL, Lv Z, Liu ZG, Hu Z, Zou GL (2007) Study on interaction between apigenin and human serum albumin by spectroscopy and molecular modeling. J Photochem Photobiol A Chem (in press)
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK Olson AJ (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19:1639–1662
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242
Wang YL, Wang HF (2002) Universitatis Pekinensia Scientiarum Naturalium 38:159–164
Lee JY, Hirose M (1999) Partially folded state of the disulfide-reduced form of human serum albumin as an intermediate for reversible denaturation. J Biol Chem 274:29303–29310
Yang P, Gao F (2002) The principle of bioinorganic chemistry. Science, Beijing
Chen YH, Yang JT, Martinez HM (1972) Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry 11:4120–4131
Eckenhoff RG, Charles E (2000) Inhaled anesthetic binding sites in human serum albumin. J Biol Chem 275:30439–30444
Qin C, Xie M-X, Liu Y (2007) Characterization of the myricetin-human serum albumin complex by spectroscopic and molecular modeling approaches. Biomacromolecules 8:2182–2189
Miller JN (1979) Recent advances in molecular luminescence analysis. Proc Anal Div Chem Soc 16:203–208
Maurice RE, Camillo AG (1981) Fluorescence quenching studies with proteins. Anal Biochem 114:199–212
Tian JN, Liu JQ, He WY, Hu ZD, Yao XJ, Cheng XG (2004) Probing the binding of scutellarin to human serum albumin by circular dichroism, fluorescence spectroscopy, FTIR, and molecular modeling method. Biomacromolecules 5:1956–1961
Chen GZ, Huang XZ (1990) The methods of fluorescence analysis, 2nd edn. Science, Beijing
Ross PD, Subramanian S (1981) Thermodynamics of protein association reactions: force contributing to stability. Biochemistry 20:3096–3102
Viviane M, Yves E, Johan H, Yvan M, Antoine V (1981) Fluorimetric analysis of binding of warfarin to human serum albumin
Karlsson CG, Rosengren AM, Andersson PO, Nicholls IA (2007) The spectrophysics of warfarin: implications for protein binding. J Phys Chem B 111:10520–10528
Sklar LA, Hudson BS, Simoni RD (1977) Conjugated polyene fatty acids as fluorescent probes: binding to bovine serum albumin. Biochemistry 16:5100–5108
Ilichev YV, Perry JL, Simon JD (2002) Interaction of ochratoxin A with human serum albumin: preferential binding of the dianion and pH effects. J Phys Chem (B) 106:452–459
Valeur B, Brochon JC (1999) New trends in fluorescence spectroscopy, 6th edn. Springer, Berlin
Zsila F, Bikadi Z, Simonyi M (2003) Probing the binding of the flavonoid, quercetin to human serum albumin by circular dichroism, electronic absorption spectroscopy and molecular modelling methods. Biochem Pharmacol 65:447–456
Xi JQ, Guo R (2007) Acid-base equilibrium of puerarin in CTAB micelles. J Pharm Biomed Anal 43:111–118
Michael D, Carter DC, Rüker F (2000) Conformational transitions of the three recombinant domains of human serum albumin depending on pH. J Biol Chem 275:3042–3050
Jamie G, Patricia ZA, Isabelle P, Bhattacharya AA, Masaki O, Stephen C (2005) Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol 353:38–52
Acknowledgements
This work is supported by the grant form National Natural Science Foundation of China (Grant No. 30370366, 30470464) and the Research Found for the Doctoral Program of Higher Education. Our great thanks to Nan Yang Zhangzhongjing Digital Chinese Medicine Co. Ltd. and Wuhan Shiji Pharmaceutical Co. Ltd. for their help.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
He, Y., Wang, Y., Tang, L. et al. Binding of Puerarin to Human Serum Albumin: A Spectroscopic Analysis and Molecular Docking. J Fluoresc 18, 433–442 (2008). https://doi.org/10.1007/s10895-007-0283-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-007-0283-0