
Abstract
This review provides an account of single-molecule fluorescence methodologies for freely diffusing molecules applied to a diverse array of biological problems pertaining to biomolecular folding and assembly. We describe the principles of confocal fluorescence microscopy to detect and analyze fluorescence bursts from diffusing single molecules. These methods including single-molecule fluorescence resonance energy transfer, coincidence, correlations and polarization offer a powerful means to uncover hidden information about conformational sub-populations and interconversion dynamics of biomolecular systems in a wide range of timescales. We offer several key examples to illustrate how these methodologies have been extremely useful in teasing out structural and dynamical aspects of many important biomolecular systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fluorescence spectroscopy has revolutionized our understanding of the structure and dynamics of complex biological systems [1–4]. The major advantages of fluorescence measurements of biological systems are (a) the inherent sensitivity of the technique, (b) a wide range of accessible timescales from femtoseconds to seconds or longer, and (c) the ability to selectively probe a sparse level of molecules of interest in a milieu of complex mixtures. A number of fluorescence observables (emission spectra, Stokes shift, intensity, quenching, polarization etc.), both in steady-state and time-resolved formats have been utilized in order to probe biomolecular structure and dynamics. In particular, fluorescence resonance energy transfer (FRET) being a spectroscopic ruler is one of the most powerful observables to track biomolecular conformational changes in the spatial range of 20–80 Å [5–7]. The sensitivity of these distance measurements arises because the FRET-efficiency, E (or the rate of energy transfer) is a function of the sixth power of the distance (r) as given by Eq. 1. The Förster’s distance (R 0, at which E = 0.5 for a given dye pair) of the dye pair in (Å) is related to the donor quantum yield (Φ D), index of refraction (η), orientation factor (κ 2) (2/3 for dynamically averaged orientation) and overlap integral (J) as given by Eq. 2 [1].
Steady-state FRET reports the change in the mean distance, while time-resolved FRET is capable of yielding intramolecular distance distributions from time-dependent fluorescence decays. For example, time-resolved FRET has proven extremely useful in demonstrating the complex conformational equilibria in protein folding reactions [8–10]. FRET experiments carried out under single-molecule resolution can provide information about structural distributions in a more direct fashion since emitted photons from individual molecules can be recorded and analyzed. This opens up an avenue to interrogate biomolecular systems in a molecule-by-molecule manner, and to extract structural and dynamical information in a more model-free fashion.
In the past few years, investigation of complex biological systems using single-molecule fluorescence methods has yielded a wealth of information that is not generally obtainable in conventional ensemble (bulk) experiments [11–20]. In addition to structural information, it singles out the dynamics of individual molecules ranging from nanoseconds to seconds depending on the observation format. Two broadly classified observation formats are described as follows: (1) Time-trajectories of fluorescence intensity from single immobilized molecules are recorded using total-internal reflection or confocal microscopy; (2) Fluorescence bursts from freely diffusing molecule through tiny observation volume are recorded using a confocal microscopy setup. The former method has been utilized in the majority of single-molecule experiments so far. The latter method is less common, but has several advantages: (1) It minimizes any extraneous factors related to protein–surface interactions arising due to immobilization. (2) data acquisition is somewhat simpler and much less time-consuming, hence one can record information on a statistically significant number of molecules within a short period of time, and (3) kinetic information under equilibrium conditions is available over a wide range of timescales from nanoseconds to milliseconds. The primary disadvantage is that since individual molecules are only probed as long as they are in the confocal volume, equilibrium kinetic process that occur between the ms and minute timescales are often more difficult to probe. Henceforth, this review will focus on single-molecule measurements on freely diffusing molecules.
From ensemble to single-molecule fluorescence measurements
In this section we provide a simplistic approach to conceptualizing the transition from ensemble (bulk) fluorescence intensity to single-molecule fluorescence bursts (Fig. 1). In conventional ensemble fluorescence experiments with steady-state excitation and under equilibrium conditions, there are no significant time-dependent changes in the emission intensity that are related to molecular fluctuations. In contrast, when the observation volume is very tiny (confocal) and the fluorophore concentration is sufficiently reduced, one observes time-dependent fluctuations in fluorescence intensity. This is due to the fact that (at typically nM concentrations) there are only a few molecules diffusing in-and-out through the observation volume giving rise to intensity fluctuations which contain information about the diffusional properties of the molecules. In the fluorescence correlation spectroscopy (FCS) approach, an autocorrelation analysis of these intensity fluctuations yields the molecular diffusion time constant, and may also contain additional faster conformational dynamics components [21–23]. If the concentration is further reduced, the relative fluctuation amplitude dramatically increases, and under this condition, one observes photon (quantum) bursts emitted from single freely diffusing molecules [24–26]. Each burst, in principle, contains information about these individual molecules. This situation is depicted schematically in Fig. 1. If the molecules are tagged with a FRET-pair (donor and acceptor), upon donor excitation, photons will be emitted from both donor and acceptor. By recording dual-color emission bursts following separation of the fluorescence by a dichroic mirror, one can measure FRET efficiencies of individual molecules [27, 28]. Multi-color detection (and excitation) with more than two spectrally resolved probes allows a powerful means to detect biomolecular association or to simultaneously measure multiple distances in molecules or complexes. The next section describes the experimental setup for data acquisition as well as data analysis used in single molecule experiments.

Schematic transition from ensemble fluorescence to single-molecule quantum bursts: Lowering concentration and squeezing down the observation volume (confocal volume, for example) gives rise to fluorescence fluctuations for in-and-out diffusion of molecules. Further lowering the concentration to ensure primarily one molecule diffusing through at a time, gives rise to fluorescence bursts. These bursts contain information about the structure and dynamics of individual molecules
Experimental setup, data acquisition and reduction
Confocal fluorescence microscopy is well-suited for data collection from diffusing single molecules [24, 28]. To do so, a collimated laser beam is focused on a sample using a high numerical aperture objective (Fig. 2a). The fluorescence is collected through the same objective, and emission light is separated from excitation light by an appropriate dichroic mirror. A 3D-gaussian observation volume (usually <1 femtoliter) is achieved by using a pinhole (10–100 μm) on the detection path (Fig. 2b). Multiple emission colors can be further separated using one or more dichroic mirrors. After filtering, the emitted photons are detected using avalanche photodiodes (APDs), which are point detectors with fast time-resolution (down to the sub-nanosecond timescale). With such a detection geometry, when the fluorophore concentration is sufficiency low (<200 pM), it is possible to detect fluorescence bursts arising from single diffusing molecules (previous section). Although these bursts are rich in information about individual molecules, several sources contribute to noise in the measurement. The most important sources include photon shot-noise (due to the low signals), fluctuations from molecular diffusion, and background counts from sources including impurities, Raman scattering and dark noise from the detectors. Often, a ratiometric approach is used in such experiments, which significantly reduces noise from sources such as diffusion [28]. For example, in single molecule FRET (SM-FRET) experiments, fluorescence bursts are recorded simultaneously in donor and acceptor channels, and FRET-efficiencies of individual molecules are estimated using a ratiometric relationship (Eq. 3).

a Experimental setup for single-molecule fluorescence measurements. For FRET measurements, photon bursts are separated from laser excitation light by a dichroic and detected by using two APDs. b Molecular diffusion through a laser-illuminated confocal volume giving rise to fluorescence bursts is schematically depicted
where I D and I A are the numbers of photon counts recorded in the donor and acceptor channels respectively, typically within a time window of a few 100 μs. Data are collected for a number of molecules (typically several hundred or more), and plotted in a histogram (number of events vs. FRET-efficiency).
In the absence of any conformational dynamics component, the resultant ratiometric FRET-efficiency profile along the burst will reflect the conformational properties of the molecule, rather than be dominated by intensity fluctuation due to the trajectory of the molecule through the confocal volume, as when only donor quenching is monitored. Because SM-FRET efficiencies are recorded on a molecule-by-molecule basis, ratiometric experiments provide a means to measure conformational subpopulations in a mixture of species. Furthermore, it is also possible to measure dynamics of conformational transitions that occur on the ca. 10 μs to 1 ms timescale (see below). In addition to FRET, polarization or spectral fluctuations are other examples of properties that can be measured in such a ratiometric fashion for single molecules.
Equation 3 assumes that the product of dye quantum yield and detection efficiency is the same for the two detection channels. In practice, to measure distances more accurately, a correction factor for both these effects is introduced as shown in Eq. 4.
where
The corrected FRET-efficiency (E corr) involves a correction factor (γ), which is dependent on the sample (the ratio of fluorescence quantum yields, ϕ A/ϕ D) and the optical geometry (the ratio of detection efficiencies, η A/η D), as given in Eq. 4. Independent measurements of quantum yield component and the ratio of the detection efficiencies allow one to obtain the γ-parameter.
More advanced data acquisition can be carried out using time-correlated single-photon counting (TCSPC) and picosecond pulsed excitation coupled with APD detection. This method can be used to classify emitted photon bursts according to fluorescence lifetime, providing another dimension (fluorescence lifetime) useful to further resolve molecular sub-populations. In its most versatile form, multi-parameter detection involves recording a number of parameters for each emitted photon, including arrival time, time delay from the excitation pulse, color and polarization. Analysis of such multiparameter data sets offers a powerful multidimensional tool to analyze the single-molecule bursts to resolve conformational heterogeneities in biomolecules and to probe several molecular properties associated with them [29]. Recently introduced two-color (donor and acceptor) alternating laser excitation methodology has been found very useful to simultaneously obtain stoichiometry and intramolecular distance in single biomolecules [30]. In addition to being capable of resolving a low-FRET population from the so-called ‘zero peak’, this method also permits more accurate distance measurements. Additionally, from the FRET-efficiency histogram, one can often delineate the dynamics of conformational interconversion for fluctuating molecules. In a system with multiple FRET-distinguishable species, three distinct cases can be envisioned: (1) conformational averaging occurs appreciably slower (or static) compared to the diffusion time (i.e. observation time), giving rise to multiple FRET-peaks corresponding to different conformations, (2) conformational exchange occurs in the range of the observation timescale, so histogram peaks show broadening, and one could extract information about the dynamics, and (3) the averaging is much faster compared to the observation time, causing the SM-FRET-distribution to show a relatively narrow (close to shot-noise limited) peak, and faster correlations FCS must be used to extract out the dynamics. All three cases have been experimentally realized in different protein and RNA systems by Deniz and coworkers (Fig. 4), as discussed in the following sections. Figure 3 summarizes the single-molecule fluorescence observables in a wide time window ranging from nanoseconds to seconds.

Different timescales and observables for single-molecule fluorescence from freely diffusing molecules
Applications to biomolecular systems
Protein folding and dynamics
The question of how an unfolded polypeptide chain assumes a uniquely folded structure has intrigued researchers over the past few decades. The current conceptual view of protein folding involves an energy landscape funnel model in which local and global minima are ascribed as intermediates and final form, respectively [31–33]. The process of folding is seemingly complex, with numerous unfolded conformations existing at the top of the funnel, each of which could fold via multiple paths and intermediates. Due to this enormous heterogeneity, it is extremely difficult to probe the folding process in detail using ensemble measurements. Single-molecule experiments offer a novel tool to probe this complexity by one molecule at a time, in principle permitting an unprecedented view of conformational populations as well as folding pathways in great detail. An early study using FRET to study protein folding at single-molecule resolution under freely diffusing conditions reported direct observations of folded and unfolded subpopulations of chymotrypsin inhibitor 2 (CI2), which varied as a function of denaturant concentration [34]. The study demonstrated in a model free manner that CI2 is a two-state folder having folded (high FRET) and unfolded (low FRET) states (Fig. 4a). Subpopulation properties were analyzed to extract a single-molecule denaturation curve, as well as a possible small expansion in the unfolded state as a function of increasing denaturant. In a subsequent study, the folding behavior of the cold-shock protein CspTm showed a similar bimodal distribution of FRET efficiencies [35]. The FRET peak corresponding to unfolded protein showed gradual shift as a function of decreasing denaturant, indicating a collapse to more compact denatured conformations at low denaturant concentration. Also, by noting that the width of the FRET efficiency distribution of denatured CspTm was very similar to that from a rigid polyproline type II helix, an upper limit for the reconfiguration time in the unfolded state was estimated to be ∼25 μs. Utilization of Kramer’s theory of chemical kinetics and stopped-flow measurements enabled the authors to estimate limits on the height of the folding free-energy barrier (Δ): 11 k B T > Δ > 4 k B T. These results clearly demonstrated that single molecule FRET experiments can provide detailed information about protein folding states and dynamics. A combined confocal microscopy setup with a microfluidic laminar mixing device also enabled the direct observation of single-molecule folding kinetics with ∼100 ms dead time [36].

Three possible kinetic scenarios of FRET-efficiency distributions. a SM-FRET histogram of a protein (chymotrypsin inhibitor 2) under moderately denaturing conditions (4M GdmCl) shows folded (high FRET) and unfolded (low-FRET) populations, frozen on the experimental timescale (modified from Deniz et al. [34]). b SM-FRET histogram of a hairpin ribozyme at 10 mM Mg2+. Peak broadening and tailing result from exchange of extended-undocked and quasi-docked conformations taking place on the experimental timescale (modified from Pljevaljčić et al. [50]). c SM-FRET histogram of the rapidly fluctuating monomeric form of a yeast prion domain showing a single peak (modified from Mukhopadhyay et al. [42])
Recent studies have aimed at further understanding the collapsed states of polypeptide chains under near-native conditions (low denaturant concentration) [37]. The collapsed state involves local structure formation in the unfolded polypeptide chain without interference from long-range Gaussian chain characteristics. The collapsed state, observed both for protein L (α/β) and CspTm (all-β), was suggested to arise from either increased inter-residue attraction or decreased excluded volume under denatured conditions [38]. The width of the FRET-efficiency histogram and a comparison with simulated shot-noise limited distributions, led to suggestions of slow (>1 ms) polypeptide chain dynamics, potentially indications of formation and dissolution of specific structures in the unfolded state. Microsecond polypeptide dynamics in unfolded proteins have also been studied using FCS, which is typically performed on a small-ensemble level with a few molecules in the confocal volume [39–41]. The method takes advantage of fluorescence intensity fluctuations due to rapid polypeptide fluctuations between quenched and un-quenched states. The rate of conformational fluctuations of unfolded intestinal fatty acid binding protein on the microsecond timescale have been directly measured [39]. These findings indicated that unfolded proteins are not simple random coils but rather form transient structures.
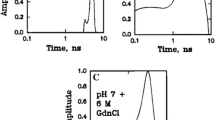
Thus, single-molecule fluorescence experiments on freely diffusing protein molecules have been extremely useful to understand several intricate aspects of the protein folding reaction. They also hold significant promise for detecting structural distribution and dynamics of natively unfolded proteins, now realized to be ubiquitous in biology. For example, single-molecule studies have been utilized very recently to study a natively unfolded and amyloid forming yeast prion protein, Sup35 [42]. The prion-determining domain (NM) of Sup35 comprises two distinct regions: an amyloidogenic Asn/Gln-rich N-terminal domain (N) and a charged solubilizing middle region (M). Using single-molecule FRET and FCS, Mukhopadhyay et al. investigated the structure and dynamics of the prion-domain under conditions where it remains monomeric. The monomeric nature of Sup35NM under these conditions was directly tested by using single-molecule fluorescence coincidence measurements. This method takes advantage of the fact that in a mixture of molecules individually labeled with either of two dyes, formation of dimers or higher order oligomers will give rise to coincident bursts in a two-color excitation/detection experiment in the same type of confocal geometry as described for SM-FRET [43, 44]. This method is powerful in detecting even a small fraction of aggregating species in the presence of monomeric species. A combination of such coincidence and SM-FRET experiments decisively established that monomeric native Sup35NM exists as a compact structure, especially interesting given the low hydrophobicity of the sequence. Additionally, the SM-FRET peak showed a progressive shift upon denaturation, indicating a lack of cooperatively formed stable structure in native Sup35NM (Fig. 5). Furthermore, FCS measurements in a pseudo cross-correlation mode, uncovered nanosecond polypeptide chain dynamics in the natively unfolded protein (Fig. 5). Taken together, these results showed that the native monomeric protein is composed of an ensemble of rapidly fluctuating collapsed structures. Thus, these single molecule and related technologies are beginning to show significant promise to uncover novel information about both natively-unfolded and amyloidogenic proteins. One significant future goal would be to extend such approaches to gain insights into the complex energy landscape of the medically very important folding/misfolding/aggregation process.

a Progressive shift in the FRET efficiency during the denaturation of the monomeric prion domain of Sup35. b Conformational fluctuation and diffusion components in the FCS autocorrelation function [42]
DNA and RNA folding and dynamics
Single molecule studies in a diffusion format have proven extremely useful in understanding the conformational dynamics in DNA molecules [25]. One biologically and biotechnologically important DNA motif is the hairpin loop, which fluctuates between open and closed conformations. Equilibrium DNA hairpin opening–closing fluctuations have been directly investigated by FCS [45]. The rate associated with the opening of the hairpin was found to be essentially independent of the characteristics of the loop, whereas the rate of closing varied significantly with the loop length and sequence. Even for such a seemingly simple DNA motif, FRET-fluctuation spectroscopy revealed sub-millisecond dynamics with a complex energy landscape between two-states in a DNA hairpin loop [46, 47]. Deviations from Arrhenius kinetics for both opening and closing of hairpin-loops were observed by FRET fluctuation spectroscopy. A recent FCS analysis has revealed a three-state mechanism for DNA hairpin folding that consists of a rapid equilibrium between open and intermediate forms and a closed form [48]. Structural distributions and dynamics of more complex DNA structures have also been studied using single molecule methods. For example, by using single-molecule FRET, two sub-populations were observed for human telomeric intramolecular quadruplex, and these were ascribed to the coexistence of parallel and anti-parallel quadruplex conformations interconverting on a minute time scale [49]. The free energy barrier in the folding energy landscape of the quadruplex has been estimated to be between 3 and 15 k B T.
Related in many ways to both the protein and DNA cases, RNA folding has emerged as one very interesting area of research in recent times. The folding energy landscape of RNA is often complicated by the presence of a number of intermediates. Like protein-based enzymes, catalytic activity in ribozymes (RNA-based enzymes) requires adoption of a correctly folded state. For the hairpin ribozyme, a systematic SM-FRET study of two-, three- and four-way junction variants characterized the role of different junction geometries and confirmed that the four-way junction strongly favors the docked (active) conformation of the two loops [50]. Interestingly, the four-way junction was able to fold to a compact structure even without the loop–loop docking interactions, and broadening of the FRET peaks (Fig. 4b) provided evidence for interconversion of extended and compact (or quasi-docked) conformations of this mimic of the undocked state. The unique stability of the four-way junction ribozyme was ascribed to the population of a native-like quasi-docked state, which reduces the entropic penalty for formation of a tertiary structure.
Other biologically relevant structural dynamics
A few additional key applications of single molecule fluorescence diffusion methods are briefly mentioned below to give a flavor for the wide range of problems being addressed by these techniques. In one study, Seidel and coworkers demonstrated directly the existence of three structurally distinct forms of HIV-reverse transcriptase-nucleic acid complexes in solution [29], providing an early example of the power of multi-parameter single molecule fluorescence detection. More recently, these methods were used to study the protein-induced folding of the U4 snRNA kink-turn [51]. The binding of the 15.5 K protein was found to induce a tightly kinked conformation in the RNA that increases the kink from 85 ± 15° to 52 ± 15°. In another study, SM-FRET was used to monitor conformational transitions of the SNARE syntaxin 1, a protein essential for exocytotic membrane fusion [52]. It was discovered that free syntaxin switches between inactive closed and active open configurations with a relaxation time of 0.8 ms. This work also pointed out the importance of regulatory factors to arrest the protein in one conformational state. SM-FRET along with correlation experiments have also been performed to investigate DNA looping by the NgoMIV restriction endonuclease [53]. At low protein concentrations, DNA loop formation was observed, whereas very high protein concentrations resulted in the formation of larger DNA aggregates mediated by DNA–protein interactions. In a different study on DNA looping also using SM-FRET, it was determined that the Lac repressor produces DNA loops with a “wrapping-away” geometry [54]. The activity and function of human telomerase have also been directly monitored using two-color fluorescence burst coincidence [55]. By analyzing the coincidence events, it has been possible to determine the number and length distribution of the products, showing the utility of this method in providing insights into the enzyme processivity. In another very innovative experiment, the rotation of proton-powered subunit in single F0F1-ATP synthase in a liposome was directly observed using diffusing single-molecule FRET experiment [56]. In these experiments, one fluorophore was attached to the rotating γ-subunit of F1 and the other to the static, nonrotating β-subunit dimer of F0. Single-molecule FRET trajectories demonstrated stepwise γ-subunit rotation during ATP synthesis and hydrolysis. In a final example, in recent SM-FRET experiments, three existing models for the initial stages of transcription were tested [57], (1) transient excursions, (2) inchworming, and (3) scrunching. Here, measurements of distances between FRET dye-pairs incorporated at specific sites within RNA-polymerase and DNA in abortively initiating transcription initiation complexes, provided evidence that initial transcription proceeds through a “scrunching” mechanism. Additionally, this work further revealed the presence of a “stressed intermediate” in initial transcription, leading the authors to postulate that accumulation of DNA-scrunching stress in the “stressed intermediate” drives abortive initiation, and also provides the driving force for promoter escape and productive initiation.
Finally, single-molecule fluorescence technology is being extended to study biologically-relevant structural dynamics in a variety of cellular processes including lipid-raft formation, cell signaling, vesicle transport, nuclear pore transport, vesicle trafficking and neuronal communication, viral infection pathways, RNA dynamics and gene expression in cells (see our review for discussion and references: Deniz et al. [59]). In particular, molecular diffusion in live cells is probed by single-fluorophore tracking or single-particle tracking that allows the dynamics of individual molecules to be tracked in real-time and detailed mechanistic questions to be probed. Such upcoming studies will enhance our ability to probe complex processes in live cells.
Conclusions and outlook
This minireview has focused on some of the major accomplishments made using fluorescence from diffusing single molecules. A key strength of the method is that conformational subpopulations and their interconversion dynamics can be analyzed for a statistically significant number of molecules within a relatively short time, while minimizing perturbations of biological molecules due to surface immobilization. With the recent advent of multicolor FRET technology for diffusing molecules, it is now possible to simultaneously monitor a number of intramolecular or intermolecular distances at a single-molecule resolution [58], adding another powerful dimension to studies of biological structure and dynamics. Using an autocorrelation analysis coupled with TCSPC, one can reliably monitor conformational dynamics ranging from nanoseconds to milliseconds. Slower dynamics can be identified from multiple FRET peaks or excessive broadening of FRET peaks. In addition to providing many detailed kinds of structural and kinetic information at single molecule resolution, diffusing experiments can also serve as a guide to plan more time consuming and involved single-molecule experiments on immobilized molecules. Various improvements from instrumentation, data analysis, probe chemistry, and more collaborative efforts from biology, chemistry and physics would immensely increase the range and quality of single-molecule fluorescence experiments [59]. Progress in selective labeling chemistry (in vitro and in vivo) will be key in extending these studies to numerous biomolecular conformational changes and function triggered by complex formation in live cells and organisms. These studies will surely shed light into many more enigmatic questions about biomolecular dynamics relevant to biosynthesis, signal transduction and the biology of disease.
References
Lakowicz JR (1999) Principles of fluorescence spectroscopy. Kluwer/Plenum, New York
Lakowicz JR (1994) Topics in fluorescence spectroscopy, vol 3. Biochemical applications. Springer, Berlin Heidelberg New York
Martin Hof RH, Fidler V (ed) (2005) Fluorescence spectroscopy in biology. Springer, Berlin Heidelberg New York
Kraayenhof R, Visser AJWG, Gerritsen HC (2002) Fluorescence spectroscopy, imaging and probes. Springer, Berlin Heidelberg New York
Stryer L (1978) Fluorescence energy-transfer as a spectroscopic ruler. Annu Rev Biochem 47:819–846
Van Der Meer BW, Coker G III, Chen S-YS (1994) Resonance energy transfer: theory and data. VCH, New York
David L, Andrews AAD (1999) Resonance energy transfer. Wiley, New York
Haas E (2005) The study of protein folding and dynamics by determination of intramolecular distance distributions and their fluctuations using ensemble and single-molecule FRET measurements. ChemPhysChem 6:858–870
Lakshmikanth GS, Sridevi K, Krishnamoorthy G, Udgaonkar JB (2001) Structure is lost incrementally during the unfolding of barstar. Nat Struct Biol 8:799–804
Lee JC, Langen R, Hummel PA, Gray HB, Winkler JR (2004) Alpha-synuclein structures from fluorescence energy-transfer kinetics: implications for the role of the protein in Parkinson’s disease. Proc Natl Acad Sci USA 101:16466–16471
Kinosita K, Yasuda R, Noji H, Ishiwata S, Yoshida M (1998) F-1-ATPase: a rotary motor made of a single molecule. Cell 93:21–24
Basche T, Moerner WE, Orrit M, Wild UP (eds) (1996) Single-molecule optical detection, imaging and spectroscopy. VCH, Weinheim
Leuba SL, Zlatanova J (eds) (2001) Biology at the single molecule level. Elsevier, Oxford
Rigler R, Orrit M, Basche T (eds) (2002) Single molecule spectroscopy. Springer, Berlin Heidelberg New York
Mehta AD, Rief M, Spudich JA, Smith DA, Simmons RM (1999) Single-molecule biomechanics with optical methods. Science 283:1689–1695
Ishijima A, Yanagida T (2001) Single molecule nanobioscience. Trends Biochem Sci 26:438–444
Weiss S (1999) Fluorescence spectroscopy of single biomolecules. Science 283:1676–1683
Bustamante C, Bryant Z, Smith SB (2003) Ten years of tension: single-molecule DNA mechanics. Nature 421:423–427
Tinnefeld P, Sauer M (2005) Branching out of single-molecule fluorescence spectroscopy: challenges for chemistry and influence on biology. Angew Chem Int Ed Engl 44:2642–2671
Michalet X, Weiss S, Jager M (2006) Single-molecule fluorescence studies of protein folding and conformational dynamics. Chem Rev 106:1785–1813
Hess ST, Huang SH, Heikal AA, Webb WW (2002) Biological and chemical applications of fluorescence correlation spectroscopy: a review. Biochemistry 41:697–705
Thompson NL (1989) Fluorescence correlation spectroscopy. In: Lakowicz JR (ed) Topics in fluorescence spectroscopy. Plenum, New York
Magde D, Elson EL, Webb WW (1974) Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 13:29–61
Eigen M, Rigler R (1994) Sorting single molecules — application to diagnostics and evolutionary biotechnology. Proc Natl Acad Sci USA 91:5740–5747
Edman L, Mets U, Rigler R (1996) Conformational transitions monitored for single molecules in solution. Proc Natl Acad Sci USA 93:6710–6715
Shera EB, Seitzinger NK, Davis LM, Keller RA, Soper SA (1990) Detection of single fluorescent molecules. Chem Phys Lett 174:553–557
Deniz AA, Dahan M, Grunwell JR, Ha TJ, Faulhaber AE, Chemla DS, Weiss S, Schultz PG (1999) Single-pair fluorescence resonance energy transfer on freely diffusing molecules: observation of forster distance dependence and subpopulations. Proc Natl Acad Sci USA 96:3670–3675
Deniz AA, Laurence TA, Dahan M, Chemla DS, Schultz PG, Weiss S (2001) Ratiometric single-molecule studies of freely diffusing biomolecules. Annu Rev Phys Chem 52:233–253
Rothwell PJ, Berger S, Kensch O, Felekyan S, Antonik M, Wohrl BM, Restle T, Goody RS, Seidel CA (2003) Multiparameter single-molecule fluorescence spectroscopy reveals heterogeneity of HIV-1 reverse transcriptase:primer/template complexes. Proc Natl Acad Sci USA 100:1655–1660
Kapanidis AN, Laurence TA, Lee NK, Margeat E, Kong XX, Weiss S (2005) Alternating-laser excitation of single molecules. Acc Chem Res 38:523–533
Bryngelson JD, Wolynes PG (1987) Spin-glasses and the statistical-mechanics of protein folding. Proc Natl Acad Sci USA 84:7524–7528
Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG (1995) Funnels, pathways, and the energy landscape of protein-folding—a synthesis. Proteins 21:167–195
Dill KA, Chan HS (1997) From Levinthal to pathways to funnels. Nat Struct Biol 4:10–19
Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S (2000) Single-molecule protein folding: diffusion fluorescence resonance energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc Natl Acad Sci USA 97:5179–5184
Schuler B, Lipman EA, Eaton WA (2002) Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature 419:743–747
Lipman EA, Schuler B, Bakajin O, Eaton WA (2003) Single-molecule measurement of protein folding kinetics. Science 301:1233–1235
Daniel N, Gopich IV, Hoffmann A, Schuler B (2007) Ultrafast dynamics of protein collapse from single-molecule photon statistics. Proc Natl Acad Sci USA 104:2655–2660
Merchant KA, Best RB, Louis JM, Gopich IV, Eaton WA (2007) Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc Natl Acad Sci USA 104:1528–1533
Chattopadhyay K, Elson EL, Frieden C (2005) The kinetics of conformational fluctuations in an unfolded protein measured by fluorescence methods. Proc Natl Acad Sci USA 102:2385–2389
Chattopadhyay K, Saffarian S, Elson EL, Frieden C (2002) Measurement of microsecond dynamic motion in the intestinal fatty acid binding protein by using fluorescence correlation spectroscopy. Proc Natl Acad Sci USA 99:14171–14176
Frieden C, Chattopadhyay K, Elson EL (2002) What fluorescence correlation spectroscopy can tell us about unfolded proteins. Adv Protein Chem 62:91–109
Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA (2007) A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci USA 104:2649–2654
Li H, Ying L, Green JJ, Balasubramanian S, Klenerman D (2003) Ultrasensitive coincidence fluorescence detection of single DNA molecules. Anal Chem 75:1664–1670
Orte A, Clarke R, Balasubramanian S, Klenerman D (2006) Determination of the fraction and stoichiometry of femtomolar levels of biomolecular complexes in an excess of monomer using single-molecule, two-color coincidence detection. Anal Chem 78:7707–7715
Bonnet G, Krichevsky O, Libchaber A (1998) Kinetics of conformational fluctuations in DNA hairpin-loops. Proc Natl Acad Sci USA 95:8602–8606
Wallace MI, Ying LM, Balasubramanian S, Klenerman D (2001) Non-Arrhenius kinetics for the loop closure of a DNA hairpin. Proc Natl Acad Sci USA 98:5584–5589
Li HT, Ren XJ, Ying LM, Balasubramanian S, Klenerman D (2004) Measuring single-molecule nucleic acid dynamics in solution by two-color filtered ratiometric fluorescence correlation spectroscopy. Proc Natl Acad Sci USA 101:14425–14430
Jung JY, Van Orden A (2006) A three-state mechanism for DNA hairpin folding characterized by multiparameter fluorescence fluctuation spectroscopy. J Am Chem Soc 128:1240–1249
Ying LM, Green JJ, Li HT, Klenerman D, Balasubramanian S (2003) Studies on the structure and dynamics of the human telomeric G quadruplex by single-molecule fluorescence resonance energy transfer. Proc Natl Acad Sci USA 100:14629–14634
Pljevaljčić G, Millar DP, Deniz AA (2004) Freely diffusing single hairpin ribozymes provide insights into the role of secondary structure and partially folded states in RNA folding. Biophys J 87:457–467
Wozniak AK, Nottrott S, Kuhn-Holsken E, Schroder GF, Grubmuller H, Luhrmann R, Seidel CAM, Oesterhelt F (2005) Detecting protein-induced folding of the U4 snRNA kink-turn by single-molecule multiparameter FRET measurements. RNA 11:1545–1554
Margittai M, Widengren J, Schweinberger E, Schroder GF, Felekyan S, Haustein E, Konig M, Fasshauer D, Grubmuller H, Jahn R, Seidel CAM (2003) Single-molecule fluorescence resonance energy transfer reveals a dynamic equilibrium between closed and open conformations of syntaxin 1. Proc Natl Acad Sci USA 100:15516–15521
Katiliene Z, Katilius E, Woodbury NW (2003) Single molecule detection of DNA looping by NgoMIV restriction endonuclease. Biophys J 84:4053–4061
Morgan MA, Okamoto K, Kahn JD, English DS (2005) Single-molecule spectroscopic determination of Lac repressor-DNA loop conformation. Biophys J 89:2588–2596
Ren XJ, Li HT, Clarke RW, Alves DA, Ying LM, Klenerman D, Balasubramanian S (2006) Analysis of human telomerase activity and function by two color single molecule coincidence fluorescence spectroscopy. J Am Chem Soc 128:4992–5000
Diez M, Zimmermann B, Börsch M, Konig M, Schweinberger E, Steigmiller S, Reuter R, Felekyan S, Kudryavtsev V, Seidel CAM, Graber P (2004) Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase. Nat Struct Mol Biol 11:135–141
Kapanidis AN, Margeat E, Ho SO, Kortkhonjia E, Weiss S, Ebright RH (2006) Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science 314:1144–1147
Clamme JP, Deniz AA (2005) Three-color single-molecule fluorescence resonance energy transfer. ChemPhysChem 6:74–77
Deniz AA, Mukhopadhyay S, Lemke EA (2007) Single-molecule biophysics: at the interface of biology, physics and chemistry. J R Soc Interface (in press) DOI 10.1098/rsif. 2007.1021
Acknowledgements
A.A.D. acknowledges support by grants R01 GM073104 and R01 GM066833 from the National Institute of General Medical Sciences/National Institutes of Health. We thank the many individuals and groups that have contributed to the data and insights presented.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mukhopadhyay, S., Deniz, A.A. Fluorescence from Diffusing Single Molecules Illuminates Biomolecular Structure and Dynamics. J Fluoresc 17, 775–783 (2007). https://doi.org/10.1007/s10895-007-0214-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-007-0214-0